
A Promising new Therapeutic Target for Heart Failure
Priti Anand1, Rongli Zhang1,2 and Puneet Anand2*
Affiliation
- 1Department of Medicine, Cardiovascular Research Institute, Case Western Reserve University, School of Medicine, Cleveland, USA
- 2Department of Medicine, Institute for Transformative Molecular Medicine, Case Western Reserve University School of Medicine, Cleveland, USA
Corresponding Author
Puneet Anand, Ph.D, Institute for Transformative Molecular Medicine, Case Western Reserve University School of Medicine, Wolstein Research Building 4107, 2103 Cornell Road, Cleveland OH 44106, USA; Tel: 216.368.5730; E-mail: pxa100@case.edu
Citation
Puneet, A. et al. A promising new therapeutic target for heart failure (2015) J Heart Cardiol 1(1): 12-13.
Copy rights
© 2015 Puneet, A. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Natriuretic-peptides; Diagnosis; Myocardial cGMP
Introduction
Editorial
Heart failure (HF)—a chronic condition defined by the inability of the heart to supply blood to the body at normal rate—affects over 5 million people in the US alone, and it is estimated that about half of the patients who develop HF die within 5 years of diagnosis[1]. During this condition, heart goes through major structural and functional changes that entail various transcriptional[2], epigenetic[3], and signaling disruptions. In particular, aberrant myocardial cGMP-mediated signaling is widely observed in patients with HF[4].
cGMP is a GTP derived cyclic nucleotide that serves as a secondary messenger for nitric oxide and natriuretic-peptide-mediated signaling pathways. cGMP is synthesized by guanylate cyclases (GCs), which are activated by natriuretic-peptides (operating on membrane bound GCs) and by nitric oxide (operating on soluble GCs)[5]. The cellular levels of cGMP are regulated by phosphodiesterases (PDEs), which catalyze the breakdown of cGMP. To date, eleven PDE families have been identified: PDEs 5, 6, and 9 exhibit specificity for cGMP, whereas the other members degrade either cAMP only or both cGMP and cAMP[5].
PDEs have unique cellular distribution, regulation, structural and functional properties that make them a very attractive target for drug development[6]. The pharmacologic inhibition of PDEs has shown great promise in the amelioration of several pathological conditions such as dementia, schizophrenia, and heart disease[6]. In addition, specific inhibition of PDE5 is used for the treatment of erectile dysfunction and benign prostatic hyperplasia[7]. Furthermore, regulation of cGMP signaling by PDE5A inhibition has been suggested as a potential new therapeutic target for chronic HF[4]. However, NO-based cGMP signaling, regulated by PDE5A, is attenuated in HF due to reduced NO bioavailability[8]. In addition, it has been reported that enhancement of cGMP levels via the NO-based pathway leads to PDE upregulation[9]. Thus, the use of PDE5A inhibition in treating HF has limitations, and warrants focusing on NO-independent signaling pathways (such as the natriuretic-peptide-based pathway) to augment the production of cGMP.
Natriuretic-peptides, atrial natriuretic-peptide (ANP), and brain natriuretic-peptide (BNP) are secreted by cardiomyocytes to lower the pressure overload by stimulating the production of cGMP via membrane bound GCs. Since the natriuretic/ cGMP-mediated signaling mechanism is disrupted in HF, it has been postulated that restoring it may have therapeutic benefits[10]. In findings reported in the March issue of Nature, Lee et al[11] studied the role of PDE9A in regulating natriuretic-peptide/ cGMP-mediated signaling in hypertrophic heart disease. In this study, the authors found an upregulation of PDE9A expression in rat neonatal cardiomyocytes in response to phenylephrine-induced hypertrophy. Analogous results were observed with both murine and human hearts in the settings of heart failure. In addition, selective inhibition of PDE9A blunted agonist-induced hypertrophy in vitro. The authors also employed a specific inhibitor of PKG (protein kinase G, which is activated by cGMP) to show that the anti-hypertrophic effects of PDE9A inhibition occur via PKG activation. Further, the authors showed that gene knockdown, or selective inhibition, of PDE9A in cardiomyocytes led to augmentation of cGMP levels in response to natriuretic peptide but not nitric oxide stimulation.
Lee et al also used mouse model of pressure overload in PDE9A knock-out animals to demonstrate an increase in cGMP levels, accompanied with attenuation of pathologic remodeling signatures such as interstitial fibrosis, cardiomyocyte hypertrophy, and pro-hypertrophic gene expression. Remarkably, the authors also demonstrate the effectiveness of PDE9A inhibition in a clinically relevant model of hypertrophy. Specifically, they found that mice with chronic PDE9A inhibition exhibited reversal of pre-established hypertrophy in a nitric oxide-independent manner. The authors also carried out unbiased phosphoproteome analyses in myocytes to reveal substantial differential phosphorylated protein pool with PDE9A versus PDE5A inhibition. Importantly, PDE9A-regulated PKG-coupled protein phosphorylation was not substantially attenuated by the inhibition of eNOS activity. This was in contrast to PDE5A and thus supports the interpretation that regulation by PDE9A of cGMP is nitric oxideindependent.
Since PDE5A regulated nitric oxide pathway is depressed in heart failure, it has been very challenging to leverage this pathway for therapeutic use, as evidenced by recent clinical trials in heart failure patients using PDE5A inhibitor[12]. The new study by Lee et al clearly demonstrates that PDE9A operates on natriuretic-peptide stimulated cGMP (and not on the nitric oxide stimulated cGMP), and that PDE9A-mediated inhibition of natriuretic-peptide/cGMP signaling is enhanced in the diseased heart (Figure), thereby supporting the notion that targeting PDE9A may have potential therapeutic benefits. As pointed out by the authors, PDE9A inhibitors appear to be well tolerated in humans; therefore, the study by Lee et al offer new possibilities for the treatment of heart failure and, most likely, other diseases.
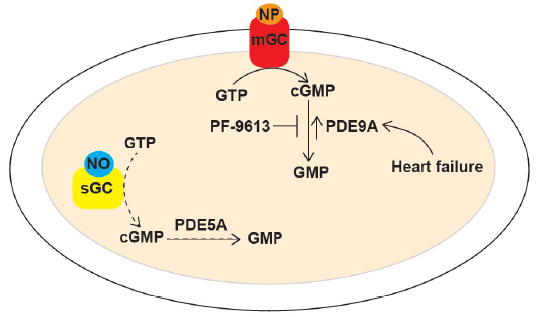
Figure 1: In the article that is subject of this editorial, Lee et al report their discovery that PDE9A regulates natriuretic-peptide/cGMP-mediated signaling in the damaged heart. In the myocardium, natriuretic-peptide (NP) or nitric oxide (NO) binds to membrane-bound guanylate cyclase (mGC) or to soluble guanylate cyclase (sGC), respectively, thereby leading to the synthesis of cGMP. Levels of natriuretic-peptide stimulated cGMP are regulated PDE9A, whereas levels of nitric-oxide stimulated cGMP are regulated by PDE5A. Nitric oxide-dependent cGMP production is depressed in cardiovascular diseases (due to reduced NO bioavailability), thereby limiting the role of PDE5A. On the other hand, PDE9A expression is upregulated during heart failure, which results in the disruption of natriuretic-peptide/cGMP-mediated signaling. Inhibition of PDE9A (by PF-9613) reverses pre-established hypertrophy by restoring this signaling pathway. Therefore, the authors propose that targeting of PDE9A may subserve a therapeutic role in heart failure and/or other heart diseases.
References
- 1. Go, A. S., Mozaffarian, D., Roger, V. L., et al. Heart disease and stroke statistics update: a report from the American Heart Association. (2013) Circulation 127(1): e6- e245.
- 2. Hill, J.A., Olson, E.N. Cardiac plasticity. (2008) N Engl J Med 358(13): 1370- 80.
- 3. Anand, P., Brown, J.D., Lin. C.Y., et al., BET bromodomains mediate transcriptional pause release in heart failure. (2013) Cell 154(3): 569- 82.
- 4. Kukreja, R.C., Salloum, F.N., Das, A. Cyclic guanosine monophosphate signaling and phosphodiesterase-5 inhibitors in cardioprotection. (2012) J Am Coll Cardiol 59(22): 1921- 1927.
- 5. Mehats, C., Andersen, C.B., Filopanti, M., et al. Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling. (2002) Trends Endocrinol Metab 13(1): 29- 35.
- 6. Jeon, Y.H., Heo, Y.S., Kim, C.M., et al. Phosphodiesterase: overview of protein structures, potential therapeutic applications and recent progress in drug development. (2005) Cell Mol Life Sci 62(11): 1198-220.
- 7. McVary, K.T., Monnig, W., Camps, J.L., et al. Sildenafil citrate improves erectile function and urinary symptoms in men with erectile dysfunction and lower urinary tract symptoms associated with benign prostatic hyperplasia: a randomized, double-blind trial. (2007) J Urol 177(3): 1071- 7.
- 8. Greene, S.J., Gheorghiade, M., Borlaug, B.A., et al. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. (2013) J Am Heart Assoc 2(6): e000536.
- 9. Mullershausen, F., Russwurm, M., Koesling, D., et al. In vivo reconstitution of the negative feedback in nitric oxide/cGMP signaling: role of phosphodiesterase type 5 phosphorylation. (2004) Mol Biol Cell 15(9): 4023- 30.
- 10. Boerrigter, G., Lapp, H., Burnett, J.C. Modulation of cGMP in heart failure: a new therapeutic paradigm. (2009) Handb Exp Pharmacol (191): 485- 506.
- 11. Lee, D.I., Zhu, G., Sasaki,T., et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. (2015) Nature 519(7544): 472- 476.
- 12. Redfield, M.M., Chen, H.H., Borlaug, B.A., et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. (2013) JAMA 309(12): 1268- 1277.










