
Alstrom Syndrome with a Mutation in Exon8 (C.4746C > A) of Alstrom Syndrome Protein 1 Gene: The First Case Report and Literature Review
Affiliation
Department of Molecular and Internal Medicine, Graduate School of Biomedical & Health Sciences, Hiroshima University, Hiroshima, Japan
Corresponding Author
Masayasu Yoneda, 1-2-3 Kasumi Minami-ku Hiroshima, Japan, 734-8551, Tel: +81-82-257-5196; Fax: +81-82-255-7360; E-mail: masayone17@hiroshima-u.ac.jp
Citation
Yoneda, M., et al. Alstrom Syndrome with a Mutation in Exon8 (C.4746C > A) of Alstrom Syndrome Protein 1 Gene: The First Case Report and Literature Review. (2017) J diab Obes 4(2): 1- 4.
Copy rights
© 2017 Yoneda, M. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Alström syndrome , hyperglycemia, type 2 diabetes, insulin resistance, liver cirrhosis
Abstract
Alström syndrome (AS) is a rare, autosomal recessive disorder characterized by progressive cone-rod dystrophy, childhood obesity, sensorineural hearing impairment, type 2 diabetes, hypogonadism, and additional abnormalities. Herein we report a 29-year-old man with progressive cone-rod dystrophy, hearing impairment, liver cirrhosis, bilateral cryptorchidism with hypergonadotropic hypogonadism, and type2 diabetes. He had severe insulin resistance and required high-dose insulin to treat his hyperglycemia. Genetic testingrevealed a mutation in exon 8 (c.4746C > A) of the Alström syndrome protein 1 (ALMS1) gene and this is the first report of this mutation in AS. It is difficult to lead to correct diagnosis without genetic test, and physicians should suspect AS when patients show early onset visual dysfunction, obesity, or type 2 diabetes.
Introduction
Alström syndrome (AS)[1] (OMIM: 203800), a very rare autosomal recessive disorder, was first reported by Alström in 1959[2]. Patients with AS presents with various symptoms, such as progressive cone-rod dystrophy, childhood obesity, sensorineural hearing impairment, type 2 diabetes, hypogonadism, short statue, and other abnormalities. AS results from a mutation in the ALMS1 gene, located on chromosome 2p13[3-5]. ALMS1 is composed of 23 exons, and it has been reported that mutation sites in patients with AS mainly exist in exons 8, 10, or 16, which account for 85% of all known sites[6]. ALMS1 is ubiquitously expressed, and the functions of ALMS1 vary in each organ. ALMS1 localizes to the centrosomes and basal bodies of ciliated cells, and is involved in intracellular signaling pathways[7-9]. Although AS is a single-gene disorder, patients with AS show different phenotypes or onsets due to not only mutation sites but also environmental cues[10].
Case Report
A 29-year-old man was followed-up in the diabetic clinic due to type 2 diabetes, liver dysfunction, and thrombocytopenia and was referred to our hospital in order to treat proceeding hyperglycemia and undergo medical detailed examinations for multi-organ dysfunction from childhood.
He was born at full-term via a normal delivery, with a birth weight of 2680 g. Photophobia was noted at the age of 1, and he gradually lost visual acuity. He completely lost his eyesight at the age of 15 due to cone-rod dystrophy. He had also presented with obesity since the age of 2, type 2 diabetes and sensorineural hearing impairment since the age of 10, and liver dysfunction and thrombocytopenia since the age of 27. He was treated with antidiabetic agents from diagnosis and his glycemic control was around HbA1c 7%.
At the age of 29, he went to a general hospital because of progressing hyperglycemia. Liver cirrhosis, splenomegaly, and an esophageal varix were subsequently diagnosed at the hospital. Initially, drug-induced hepatitis was suspected; hence, all antidiabetic agents (metformin, glimepiride, and voglibose) were stopped, and insulin therapy with 4 injections/day was initiated. Although he used more than 100 U/day of insulin, his glycemic control remained poor. He also had diabetic complications; diabetic kidney disease, diabetic retinopathy and neuropathy. In addition, hypergonadotropic hypogonadism was identified. He was referred to our hospital for hyperglycemia treatment and undergoing medical detailed examinations for multi-organ dysfunction.
The patient’s parents were fifth-degree relatives; hence, we suspected some genetic disorders (figure 1). He had a healthy, unaffected older brother, and no relatives possessed similar diseases.
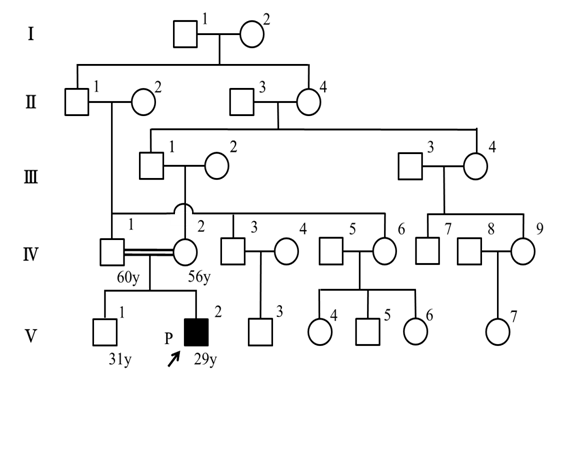
Figure 1: The pedigree of our AS patient. The case (proband) is indicated by a black square. His paternal grandfather and his maternal great-grandmother were sibling.
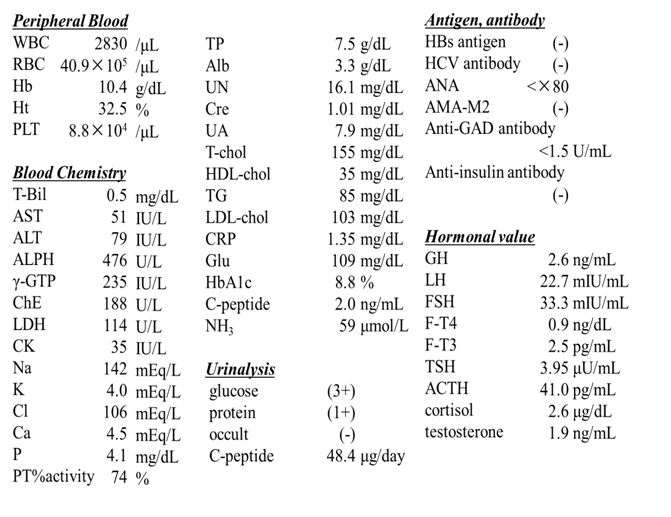
On a physical examination in our hospital, he was 154.5 cm in height, 46.0 kg in weight, and his body mass index was 19.3 kg/m². Body temperature was 36.3°C. He reported no alcohol consumption. Bilateral cryptorchidism was evident in the absence of polydactyly, neurological dysfunction, and skin tags. No splenomegaly, jaundice, or edemas were observed. Laboratory data (Table 1) showed elevated liver enzyme levels, pancytopenia, and hypoalbuminemia. He was negative for hepatitis B surface (HBs) antigen, hepatitis C virus (HCV) antibody, and autoantibodies [anti-nuclear antibody (ANA) and anti-mitochondrial M2 antibody (AMA-M2)]. Glycemic control was poor (HbA1c 8.8%). CRP elevation was likely due to aspiration revealed in the swallowing test. Hypergonadotropic hypogonadism and cryptorchidism were also observed. Heptic atrophy, hepatosteatosis, splenomegaly, esophageal varix, and stomach ulcer were identified by contrast-enhanced computed tomography (CT) and upper gastrointestinal endoscopy. Funduscopy revealed cone-rod dystrophy. Audiometry revealed bilateral sensorineural hearing impairment. Ohwaki’s intelligence test showed no intellectual disturbance or mental developmental delay.
Table 1: Laboratory data.
Insulin dose was titrated upwards to (3.0 U/kg) with concurrent diet (1500 kcal, protein 50 g/day) and exercise therapy, resulting in agradual improvement in glycemic control by the fourteenth hospital day (Figure 2) and was discharged from our hospital on the twenty-eighth hospital day. Liver biopsy was not performed, because he could not follow instructions due to the hearing impairment. Testosterone replacement therapy was not pursued because he had developed secondary sexual characteristics and asymptomatic upon physical examination.
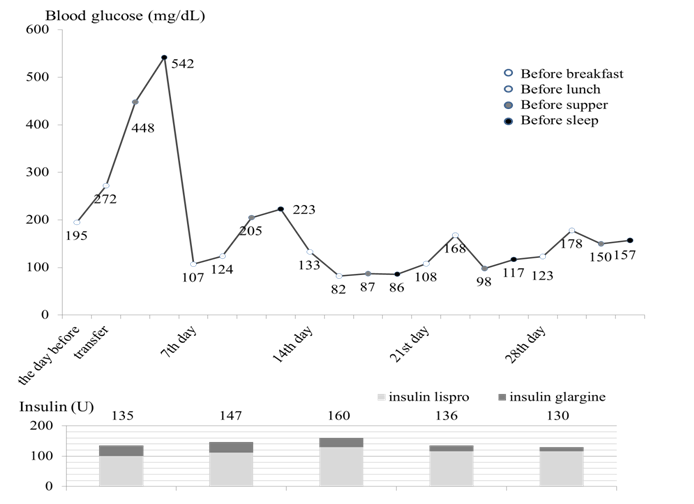
Figure 2: Clinical course of this case. The blood glucose levels were controlled around 150 mg/dL with a total of 130 U per day (3.0 U/kg/day) of insulin.
We suspected some genetic disorder, especially AS, because of multi-organ dysfunction. We requested a genetic analysis to Dr. Marshall, identified a single nucleotide substitution at position 4746[6] (c.4746C > A in exon 8) and finally diagnosed AS. We obtained written informed consent for the genetic analysis from the patient and his family. This study was approved by the Hiroshima University ethical committee.
Discussion
Bardet-Biedl Syndrome (BBS) and Laurence-Moon Syndrome (LMS) are disorders related to AS that share the same symptoms of AS, including low vision, hearing loss, type 2 diabetes, and liver dysfunction; hence, it is sometimes difficult to distinguish these syndromes from AS. This patient had no paraplegia and no mental retardation, the main criteria for the diagnosis of LMS. He exhibited clinical diagnostic criteria of BBS, but had no polydactyly and no intellectual disturbance, characteristic symptoms of BBS. He also exhibited clinical diagnostic criteria of AS[11], one major criteria (low vision) and five minor criteria (insulin resistance and type 2 diabetes mellitus, hearing loss, hepatic dysfunction, short stature and hypogonadism). Therefore, we suspected AS, and an ALMS1 mutation was identified by genetic testing.
AS is an autosomal recessive disorder, and the prevalence of AS is estimated as less than 0.001%[1]. AS results from mutations in the ALMS1 gene located on chromosome 2p13[7]. The ALMS1 gene contains 23 exons, and 85% of mutations are present in exons 8, 10, and 16[6]. To date, 239 different mutations have been described in ALMS1[6]. The same mutation found in our patient has been reported in only one case[6], and this is the first detailed case report of this mutation.
ALMS1 mRNA is ubiquitously expressed in the brain, lung, and testis. The ALMS1 protein has diverse roles in each organ[10]. It has been reported that patients with mutations in exon 16, where most mutations have been found, have more severe disease phenotypes, and that patients with mutations in exon 8 have a lower incidence of renal dysfunction[12].
It has been reported that 70% of AS patients have type 2 diabetes by 20 years of age, with a median age of onset of 16 years, and have severe insulin resistance[12]. In our patient, 3.0 U/kg of insulin was required to improve glycemic control, because of his severe insulin resistance. Patients with AS often have much more severe insulin resistance than controls, even when matched for age and body composition[13]. Insulin resistance was possibly caused by hepatosteatosis or silent liver damage, obesity, and attenuation of insulin signals by aging. In addition, more than half of female AS patients demonstrate various signs of polycystic ovary syndrome (PCOS) or hyperandrogenism[14]. Hence, obesity caused by those diseases sometimes contributes insulin resistance[15,16]. In our patient, severe insulin resistance was supposed to becaused by liver cirrhosis and aspiration pneumonia. Several studies demonstrated that metformin and thiazolidine, insulin-sensitizers, and exenatide, a glucagon-like peptide-1 (GLP-1) analogue, were effective for the treatment of diabetes in patients with AS[14,17,18]. In our patient, isolated insulin therapy was applied because of liver cirrhosis.
There are various phenotypes of liver dysfunction in patients with AS. Liver dysfunction begins with clinically silent elevation of transaminases and hepatosteatosis, and some patients can progress to liver fibrosis or cirrhosis[19-23]. Inflammatory changes resulting in fibrosis do not appear to be autoimmune related, because patients test negative for antinuclear antibodies and other typical markers of autoimmune hepatitis[24]. Phenotypic variation of disease progression in AS patients carrying the same mutation suggests that there might be some interplay between multiple potential genetic modifiers and environmental or infectious exposures. In our patient, inspite of cirrhosis suspected by hepatic atrophy and esophageal varix identified by CT and upper gastrointestinal endoscopy, the cause of cirrhosis was not revealed since liver biopsy was not pursued. He had silent elevation of transaminases since the age of 27, so the best etiologic evidence of his liver cirrhosis was the presence of elevated transaminases and hepatosteatosis.
Although AS is very rare, physicians should suspect AS when patients show early onset visual dysfunction, obesity, or type 2 diabetes. We believe that early diagnosis may provide better medical treatment, lengthen survival, and improve quality of life.
Acknowledge:
We thank Dr. J. D. Marshall, Genetics Coordinator Alstrom Syndröme Studies, The Jackson Laboratory, Bar Harbor, USA. The authors state that they have no Conflict of Interest (COI).
References
- 1. Orphanet
- 2. Alstrom, C.H., Hallgren, B., Nilsson, L.B., et al. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. (1959) Acta Psychiatr Neurol Scand Suppl 34 (129): 1-35
- 3. Collin, G.B., Marshall, J.D., Cardon, L.R., et al. Homozygosity mapping of Alstrom syndrome to chromosome 2p. (1997) Hum Mol Genet 6(2): 213–219.
- 4. Collin GB, Marshall JD, Boerkoel CF, Levin AV, Weksberg R, Greenberg J, Michaud JL, Naggert JK, Nishina PM.. Alström syndrome: further evidence for linkage to human chromosome 2p13. (1999) Hum Genet 105(5): 474–479.
- 5. Macari, F., Lautier, C., Girardet, A., et al. Refinement of genetic localization of the Alstrom syndrome on chromosome 2p12-13 by linkage analysis in a North African family. (1998) Hum Genet 103(6): 658–661.
- 6. Marshall, J.D., Muller, J., Collin, G.B., et al. Alström syndrome: mutation spectrum of ALMS1. (2015) Hum Mutat 36(7): 660–668.
- 7. Andersen, J.S., Wilkinson, C.J., Mayor, T. Proteomic characterization of the human centrosome by protein correlation profiling. (2003) Nature 426(6966): 570–574.
- 8. Hearn, T., Renforth, G.L., Spalluto, C., et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. (2003) Nat Genet 31(1): 79-83.
- 9. Hearn, T., Spalluto, C., Phillips, V.J., et al. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the Pathogenesis of Obesity, Insulin Resistance, and Type 2 Diabetes. (2005) Diabetes 54(5): 1581–1587.
- 10. Collin, G.B., Marshall, J.D., Ikeda, A., et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. (2002) Nat Genet 31(1): 74-78.
- 11. Marshall, J.D., Elizabeth, G., Hinman, E.G., et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. (2007) Hum Mutat 28(11): 1114-1123.
- 12. Marshall, J.D., Beck, S., Maffei, P., et al. Alström syndrome. (2007) Eur J Hum Genet 15(12): 1193-1202.
- 13. Minton, J.A., Owen, K.R., Ricketts, C.J., et al. Syndromic obesity and diabetes: Changes in body composition with age and mutation analysis of ALMS1 in 12 United Kingdom kindreds with Alström syndrome. (2006) J Clin Endocrinol Metab 91(18): 3110–3116.
- 14. Sinha, S.K., Bhangoo, A., Anhalt, H., et al. Effect of metformin and rosiglitazone in a prepubertal boy with Alstrom syndrome. (2007) J Pediatr Endocrinol Metab 20(9): 1045–1052
- 15. Poretsky, L., Cataldo, N.A., Rosenwaks, Z., et al. The insulin-related ovarian regulatory system in health and disease. (1999) Endocr Rev 20: 535-582.
- 16. Diamanti-Kandarakis, E., Dunaif, A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. (2012) Endocr Rev 33(6): 981–1030.
- 17. Girard, D., Petrovsky, N. Alström syndrome: insights into the pathogenesis of metabolic disorders. (2011) Nat Rev Endocrinol 7(2): 77-88.
- 18. Paisey, R.B. New insights and therapies for the metabolic consequences of Alstrom syndrome. (2009) Curr Opin Lipidol 20(5): 315–320.
- 19. Izzi, C., Maffei, P., Milan, G., et al. The CaseFamilial occurrence of retinitis pigmentosa, deafness, and nephropathy. (2011) Kidney Int 79(6): 691–692.
- 20. Marshall, J.D., Maffei, P., Collin, G.B., et al. Alstrom syndrome: genetics and clinical overview. (2011) Curr Genomics 12(3): 225–235.
- 21. Connolly, M.B., Jan, J.E., Cough, R.M., et al. Hepatic dysfunction in Alströmdisese. Am J Med Genet 40(4): 421-424
- 22. Awazu, M., Tanaka, T., Sato, S., et al. Hepatic dysfunction in two sibs with Alström syndrome: Case report and review of the literature. (1997) Am J Med Genet 69(1): 13-16
- 23. Quiros-Tejeira, R.E., Vargas, J., Ament, M.E. Early-onset liver disease complicated with acute liver failure in Alström syndrome. (2001) Am J Med Genet 101(1): 9-11.
- 24. Marshall, J.D., Bronson, R.T., Collin, G.B., et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. (2005) Arch Intern Med 165(6): 675–683.










