
Assessment of Mutation Genetics in HTT (Hi-CAG), Gene for Induced Huntingtonâs disease in Tabriz, Iran
Shahin Asadi1*, Zahra Gholizadeh1, Maryam Safi2, Mina Niknia2, Mahsa Jamali2
Affiliation
- 1Research Center for Stem Cell and Drug Applied Research Center, Tabriz University of Medical Sciences in modern biology, Iran
- 2Young Researchers and Elite Club,Tabriz Branch, Islamic Azad University, Tabriz, Iran
Corresponding Author
Shahin Asadi, Research Center for Stem Cell and Drug Applied Research Center, Tabriz University of Medical Sciences in modern biology, Iran, Tel: +989379923364; E-mail: shahin.asadi1985@gmail.com
Citation
Asadi, S., et al. Assessment of Mutation Genetics in HTT (Hi-CAG), Gene for Induced Huntington’s disease in Tabriz, Iran. (2017) Cell Immunol Serum Biol 3(2): 93- 98.
Copy rights
© 2017 Asadi, S. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Genetic study; HuntingtonÃĸâŦâĸs disease (HD); Mutation The gene HTT
Abstract
In this study we have analyzed 120 people. 59 Huntington’s disease (HD) and 61 control group. The gene HTT analyzed in terms of genetic mutation made. In this study, people who have genetic mutation were targeted, with nervous disorders, Huntington’s disease (HD). In fact, of all people with Huntington’s disease (HD), 59 Huntington’s disease (HD) had a genetic mutation in the gene HTT Huntington’s disease (HD). Any genetic mutations in the target genes control group, did not show.
Introduction
Today, neurological disorders, neuromuscular disorders are very important in creating. Including neurological disorders, including Huntington’s disease (HD). Huntington’s disease (HD) is a neuromuscular disorder that commonly causes A progressive neurodegenerative disease with autosomal dominant inheritance, with the incidence in adulthood, the disease has three abnormal movements, cognitive disorders, psychological disorders known.. Huntington’s disease (HD) is caused by genetic mutations, but also epigenetic factors are critical in inducing the disease.
Huntington’s disease (huntington) is a genetic disease caused by defects in the gene and chromosome 4 caused. Huntington’s is an inherited disease that damages the nerve cells in the brain. Most people who have Huntington’s disease, symptoms of the disease at the age of 40 and 50 years, respectively. But sometimes sooner or later the age of onset of symptoms. If symptoms occur earlier than 20 years, it is called juvenile Huntington[1]. If we Huntington’s disease based on age classification, will be divided into two categories:
The first: The most common symptoms of Huntington’s disease after the age of 40 and 50 years old[2].
The second category: a few are infected in their teens and early onset of symptoms.
If one parent has Huntington’s disease, Huntington’s likely that their children will be affected, 50 percent[3].
If a person does not receive the defective gene from their parents, it could pass the gene to their children.
But if a child who has one parent has Huntington, Huntington’s gene, they are not; it does not transmit to their children[4].
Symptoms of Huntington’s disease
Movement Disorders: It can be both voluntary movements and involuntary movements will be included. Impairment of voluntary movements may have a greater impact on the ability of the individual and his relations with others interfere[5,6]. Movement disorders, including the following cases:
Jerking
Contraction involuntary and sustained muscle (dystonia)
Muscle rigidity
Slow and uncoordinated movements
Abnormal eye movements slow
Difficulty walking and balance
Speech disorder
Swallowing Food
Cognitive Disorders:
Inability to start or conversation
Disruption to prioritize tasks, and organizing sub program
Lack of flexibility
Lack of impulse control, which could lead to flooding desires.
Act without thinking and moral issues and sexual problems[7,8].
Impaired perception of space that can be used to take the lead so that if a person is clumsy.
Difficulty concentrating on one task for a long time
Slowness in processing thoughts and find the words
Difficulty and impairment in learning new information.
Psychiatric Disorders: The most important and most common mental disorders in Huntington’s disease, depression. Symptoms of depression include:
Feel grief
Loss of interest in normal daily activities and
Insomnia or sleeping too much
Severe fatigue and loss of energy
Feelings of worthlessness and guilt
Disruption of the decision, distractibility and decreased concentration
Thoughts that may lead to suicide
Changes in appetite
Reduction or change in sexual desire, sexual behavior inappropriate.
Other mental disorders: Obsessive-compulsive disorder: intrusive thoughts and repetitive behaviors
Mania or manic mood that is bad temper
Lack of motivation and lack of interest
Anxiety
Symptoms of Huntington’s disease in adolescence
Initiation and progression of Huntington’s disease in adolescents and young people is a little different than adults. Problems such as:
The loss of what has already been learned and the skills that person has earned
Quick academic failure behavioral problems
Stiff and rigid muscles in children with Huntington’s walk is evident
Changes in motor skills such as handwriting
The latest tremor and involuntary movements Seizure
Treatment of Huntington’s disease
There is no cure for this disease. The sole purpose of medical treatment, slowing the disease and its symptoms[9-14]. Prevention of Huntington’s disease
Genetic counseling is the best way to prevent the disease.
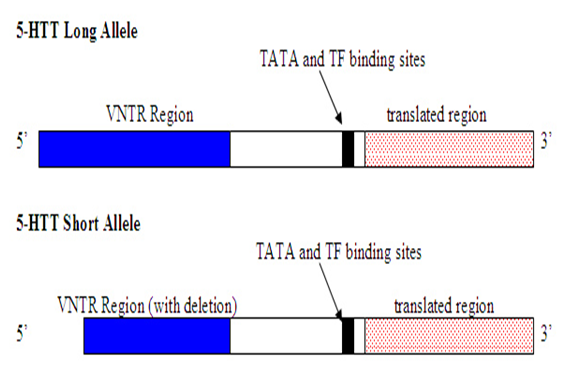
Figure 1: Schematic view of the HTT gene with long and short alleles.
Materials and Methods
In this study, 59 patients with Huntington’s disease (HD), and 60 control groups were studied. Peripheral blood samples from patients and parents with written permission control were prepared. After separation of serum, using Real Time-PCR technique of tRNA molecules were collected. To isolate Neuroglial cells erythrocytes were precipitated from hydroxyethyl starch (HES) was used. At this stage, HES solution in ratio of 1 to 5 with the peripheral blood of patients and controls were mixed. After 60 minutes of incubation at room temperature, the supernatant was removed and centrifuged for 17 min at 450 Gera. The cell sediment with PBS (phosphate buffered saline), pipetazh and slowly soluble carbohydrate ratio of 1 to 2 on ficole (Ficol) was poured in the 780G was centrifuged for 24 minutes. Mono nuclear Neuroglial cells also are included, has a lower density than ficole and soon which they are based. The remaining erythrocytes have a molecular weight greater than ficole and deposited in test tubes.
The supernatant, which contained the mono nuclear cells, was removed, and the 400 Gera was centrifuged for 19 minutes. Finally, the sediment cell, the antibody and Neuroglial cells was added after 34 minutes incubation at 5°C, the cell mixture was passed from pillar LSMACS. Then the cells were washed with PBS and attached to the column LSMACSS pam Stem cell culture medium containing the transcription gene HTT, and were kept.
To determine the purity of Neuroglial cells are extracted, flow cytometry was used. For this purpose, approximately 4 - 5 × 103 Neuroglial cells were transferred to1.5 ml Eppendorf tube and then were centrifuged at 2000 rpm for 7 minutes at time. Remove the supernatant culture medium and there mining sediment, 100 μl of PBS buffer was added. After adding 5 - 10 μl PE monoclonal anti body to the cell suspension for 60 min at 4°C incubated and read immediately by flow cytometry. For example, rather than control anti body Neuroglial cells PE, IgG1 negative control solution was used.
Total mRNA extraction procedure includes:
1 ml solution spilled Qiazolon cells, and slowly and carefully mixed and incubated at room temperature for 5 minutes. Then 200 μl chloroform solutions to target mix, and then transfer the micro tubes were added, and the shaker well was mixed for 15 seconds. The present mix for 4 minutes at room temperature and then incubated for 20 min at 4°C and was centrifuged at 13200 rpm era. Remove the upper phase product was transferred to a new microtube and to the one times the volume of cold ethanol was added. The resulting mixtures for 24 hours at -20°C were incubated.
Then for 45 min at 4°C an was centrifuged at 12000 rpm era. Remove the supernatant and the white precipitate, 1 ml of cold 75% ethanol was added to separate the sediment from micro tubes were vortex well. The resulting mixture for 20 min at 4°C and by the time we were centrifuged 12000 rpm. Ethanol and the sediment was removed and placed at room temperature until completely dry deposition. The precipitate was dissolved in 20 μl sterile water and at a later stage, the concentration of extracted mRNA was determined.
To assessment the quality of mi-RNAs,the Real Time-PCR technique was used. The cDNA synthesis in reverse transcription reaction (RT) kit (Fermentas K1622) and 1 μl oligoprimers 18 (dT) was performed. Following the PCR reaction 2 ΜM dNTP, 1 μg cDNA, Fermentas PCR buffer 1X, 0 / 75 μM MgCl2, 1.25 U/μL Tag DNA at 95°C for 4 min, 95°C for 30s, annealing temperature 58°C for 30s, and 72°C for 30 seconds, 35 cycles were performed. Then 1.5% agarose gel, the PCR product was dumped in wells after electrophores is with ethidium bromide staining and color was evaluated.
Figure 2: Microscopic view of HTT gene and brain imaging in patients with Huntington’s statistical charts.
Figure 3: Schematic view of the pattern formed in the band HTT gene with genetic mutations with chart diagram of the in patients Huntington’s and control group.
Figure 4: Schematic view of the pattern formed HTT gene and immunoglobulin gamma-band frequency diagram charts with mutations in the HTT gene in Huntington’s patients and the control group.
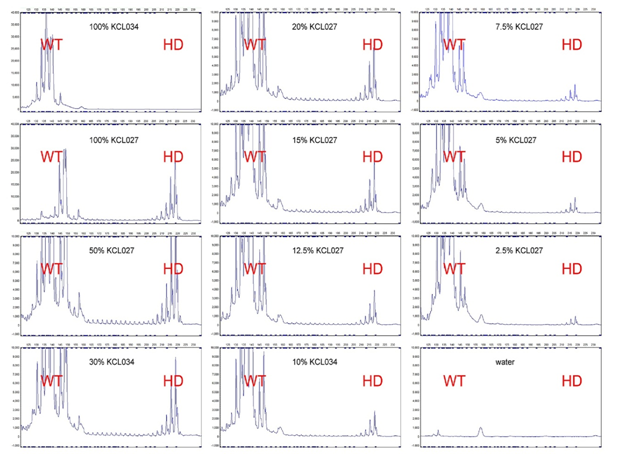
Figure 5: Schematic view of the EEG in patients with Huntington’s disease with HTT gene mutation compared with the control group.
Discussion and Conclusion
According to the results of sequencing the genome of patients with Huntington’s disease (HD), and the genetic mutation HTT gene found that about 99% of patients with Huntington’s disease (HD), they have these genetic mutations. Patients with Huntington’s disease (HD), unusual and frightening images in the process of Huntington’s disease (HD), experience. Lot epigenetic factors involved in Huntington’s disease (HD). But the most prominent factor to induce Huntington’s disease (HD), mutation is HTT gene. This gene can induce the birth and can also be induced in the adulthood.
Acknowledge:
Thanks to everyone who helped us in doing this project, very grateful. Patients and their families also accept patients who had very much for your cooperation in this study.
References
- 1. Dayalu, P., Albin, R.L. Huntington disease: pathogenesis and treatment. (2015) Neurol Clin 33 (1): 101-114.
- 2. Warby, S.C., Graham, R.K., Hayden, M.R. Huntington Disease. (2014).
- 3. Durr, A., Gargiulo, M., Feingold, J. The presymptomatic phase of Huntington disease. (2012) Rev Neurol 168(11): 806-808.
- 4. Van Duijn, E., Kingma, E.M., Van der Mast, R.C. Psychopathology in verified Huntington's disease gene carriers. (2007) J Neuropsychiatry Clin Neurosci 19(4): 441-448.
- 5. Kremer, B. Clinical neurology of Huntington's disease. (2002) Oxford University: 28-53.
- 6. Wagle, A.C., Wagle, S.A., Marková, I.S., et al. Psychiatric Morbidity in Huntington's disease. (2000) Neurol, Psychiatry and Brain Res (8): 5-16.
- 7. Aziz, N.A., Van der Marck, M.A., Pijl, H., et al. Weight loss in neurodegenerative disorders. (2008) J Neurol 255 (12): 1872-1880.
- 8. Gagnon, J.F., Petit, D., Latreille, V., et al. Neurobiology of sleep disturbances in neurodegenerative disorders. (2008) Curr Pharm Des 14(32): 3430-3445.
- 9. Murray, E.D., Buttner, E.A., Price, B.H. Depression and Psychosis in Neurological Practice. (2012) Neurol Clinical Practice: 102-103.
- 10. Semaka, A., Creighton, S., Warby, S., et al. Predictive testing for Huntington disease: interpretation and significance of intermediate alleles. (2006) Clin Genet 70(4): 283-294.
- 11. Cooper, G., Eichhorn, G., Rodnitzky, R.L. Parkinson's disease: Neuroscience in medicine. (2008) Humana Press: 508-512.
- 12. Rodriguez-Oroz, M.C., Jahanshahi, M., Krack, P., et al. Initial clinical manifestations of Parkinson's disease: features and pathophysiological mechanisms. (2009) Lancet Neurol 8(12): 1128-1139.
- 13. Fung, V.S., Thompson, P.D. Parkinson's disease and movement disorders. (2007) Rigidity and spasticity:504-513.
- 14. Yao, S.C., Hart, A.D., Terzella, M. An evidence-based osteopathic approach to Parkinson disease. (2013) Osteopathic Family Physician 5(3): 96-101.










