
Design, Modeling, Synthesis and In Vitro Evaluation of new DPP-IV Inhibitors
Affiliation
Department of Pharmaceutical Sciences, School of Pharmacy, MCPHS University, 179 Longwood Avenue, Boston Ma 02115 USA.
Corresponding Author
Department of Pharmaceutical Sciences, School of Pharmacy, MCPHS University, 179 Longwood Avenue, Boston Ma 02115 USA, Tel:++617-732-2955; E-mail: Ahmed.mehanna@mcphs.edu
Citation
Mehanna, A.S. Design, Modeling, Synthesis and in Vitro Evaluation of New DPP-IV Inhibitors. (2018) J Med Chem Toxicol 3(1): 100- 110.
Copy rights
© 2018 Mehanna, A.S. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License
Keywords
Sitagliptin; DPP-IV inhibitors; antidiabetic agents
Abstract
Dipeptidyl peptidase-IV enzyme is one of the targets for drug design to compact type-2 diabetes. The current research addresses the design, modeling, synthesis and in vitro testing of potential new DPP-IV inhibitors. X-ray structure of Sitagliptin with DPP-IV indicated hydrophobic interaction of the triazolo-piperazine ring system with the S2 pocket through the amino acid Phe357. Using molecular modeling; we designed new compounds with better hydrophobic properties facing Phe357. Six compounds were designed, docked, synthesized and tested for DPP-IV inhibitory activity. All designed compounds showed comparable affinity to the catalytic site of the enzyme to that of the standard drug, however, the activity as inhibitors, as reflected by the IC50 range of 0.3 μM – 1.3 μM, was lower than that of Sitagliptin (IC50 22 nM). The most active compound in the new series with, IC50 of 0.3 μM, showed interesting a flipped over docking mode that is different from all other compounds including Sitagliptin with the lipophilic area directed away from the amino acid Phe357. The forces of interaction of the most active compound with the enzyme catalytic site were determined from modeling studies, binding can be optimized by further structural modifications.
Introduction
DPPIV inhibitors are relatively new class of drugs introduced to the market for the treatment of type2 diabetes. Inhibition of DPP-IV results in prolonging the half-life of the glucagon-like peptide (GLP-1). The latter, is a polypeptide hormone secreted by L-cells of the gastric mucosa in response to the presence of nutrients in the lumen of small intestine to induce insulin secretion from the pancreas[1,2]. GLP-1 has a very short half-life of 2 minutes due to the rapid deactivation by DPP-IV enzyme. Accordingly, development of inhibitors for DPP-IV was found to be effective in prolonging the GLP-1 half-life to several hours. Several inhibitors were developed and successfully introduced to the market as a novel drug class to treat type-2 diabetes. Sitagliptin, developed by Merck Co[3] was the first of those DPP -IV inhibitors to hit the market for clinical use. Following Sitagliptin, several other DPPP -IV inhibitors were developed and approved by the FDA for clinical use[4] the structures of Sitagliptin and clinical approved inhibitors are depicted in (Figure-1). The inhibitors appear to have no common structural features or pharmacophore. Each inhibitor has its own unique parent nucleus and different pharmacophores. The current research was directed towards Sitagliptin, the prototype DPP -IV inhibitor. The chemical structure of the drug depicts two characteristic pharmacophores; the β-amino -amide and the triazolopiperazi ne ring system. The SAR of Sitagliptin was extensively studied by Merck scientists[5].
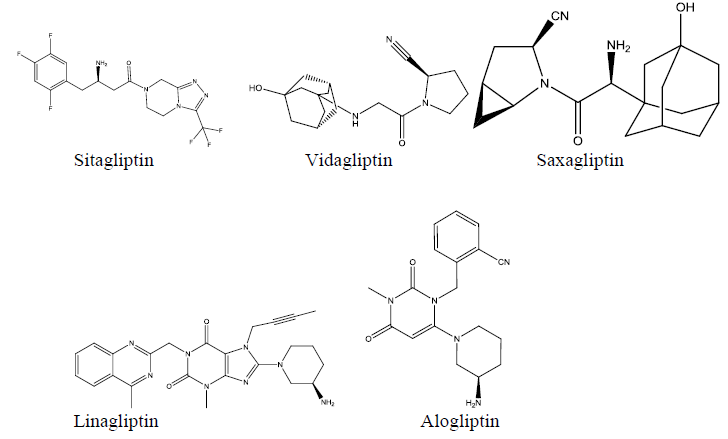
Figure 1: Commercially available DPP-IV inhibitors.
Rationale of design
X-ray crystal structure of Sitagliptin bound to DPP-IV, (Figure-2), illustrates that the triazolo-piperazine fragment of the drug is oriented towards the Phe357 residue in the catalytic site, and the trifluromethyl group towards the Ser209 residue. Both residues are part of what is known as the S2 pocket of the catalytic site.
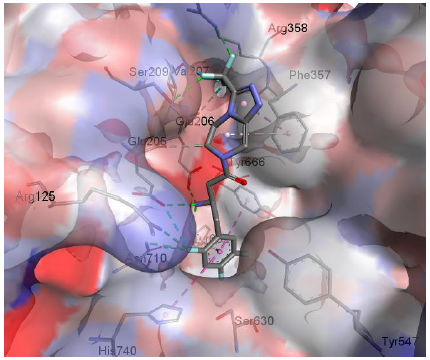
Figure 2: Sitagliptin in the DPP-IV active site (from crystal structure).
The current research aimed to design molecules maintaining the β-amino amide pharmacophore of Sitagliptin, but with the triazolo-piperazine ring system replaced by more lipophilic entities for better hydrophobic interactions with Phe 357 in the S2 pocket. (Figure 3), depicts the chemical structures of the designed molecules. Guided by molecular modeling, the six designed compounds were docked into DPP-IV catalytic sites to identify their binding poses and compare it with the standard drug pose in the crystal structure. Sitagliptin was also subjected to the same process and energy minimization and superimposition over the crystal structure form to validate the docking process. Compounds 8 and 9 differ in the number of carbons in the linker between the aromatic ring and the β-amino-amide functionality. Same applies for compounds 10 and 12 with the two methoxy groups. Compound 11, on the other hand, with fluro-substituent, designed to test the effect of electron withdrawing group on binding mode and activity, if any. Compound 13 (the phenylalanine derivative) differs from other five compounds shown in figure 3 in having a carboxylic acid group in the side chain. The latter, was designed to reach out to Arg358 observed in the active site of the S2 pocket for binding enhancement.
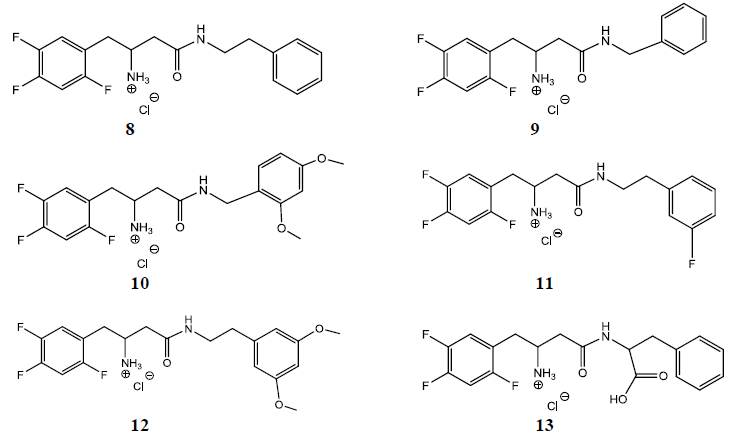
Figure 3: Chemical structures of designed molecules.
Chemistry
The synthetic pathway of the target molecules and its intermediates is outlined in scheme 1. All molecules were synthesized from intermediate 7. The synthetic steps for the latter, involved the coupling of 2-(2, 4, 5-triflourophenyl) acetic acid 1 with 2, 2-dimethyl-1, 3-dioxane-4, 6-dione (Meldrum’s acid)[6] using the activating agent trimethylacetylchloride (Pivaloylchloride) to produce coupling product 2. Reflux of 2 with a mixture of methanol and toluene yielded the β-Keto-esters 3. Reflux of 3 with ammonium acetate/methanol yielded the β-enamino ester 4. The latter was converted to 5 by reduction with sodium boro hydride in presence of acetic acid[7]. The β-amino group of 5 was protected as Boc using Boc-anhydride to give compound 6. Subsequent hydrolysis of the methyl ester yielded the common intermediate 7. The latter, with N-Boc as protecting group, and using DEPBT [3 - (Diethoxyphosphoryloxy) - (1, 2, 3) –benzotriazin - 4 (3H) - one] is coupled[8,9] with the corresponding amines, or phenylalanine methyl ester (in compound 13i), to give intermediate compounds 8a-13a. The target compounds, 8-13, were obtained as hydrochloride salts by removing Boc from 8a-13a with 4M HCl in dioxane/water[10]. See experimental part for details on the synthesis for all intermediates and final products.
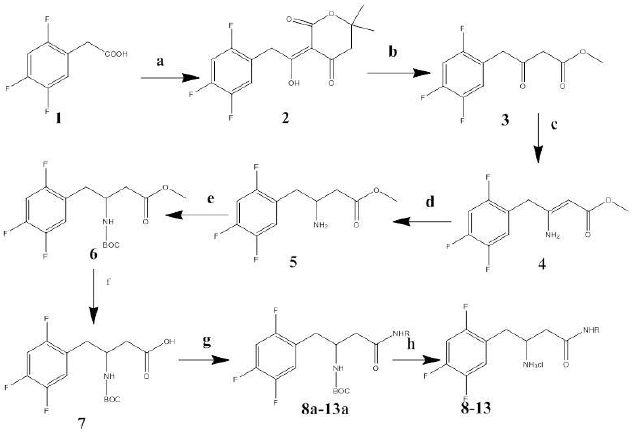
R= phenylethyl (8), benzyl (9), 2, 4-dimethoxy benzyl (10), 3-fluro-phenylethyl (11), 3, 5-dimethoxyphenylethyl (12), phenyl-ethyl-α-methyl-carboxylate (13).
(a) Meldrum’s Acid, DMAP, Acetonitrile, N, N- Diisopropylethylamine, Pivolyl chloride, 45°C
(b) Methanol, Toluene, Reflux 3 hrs
(c) Ammonium Acetate, Methanol , Reflux 7 - 8 hrs
(d) Sodium borohydride, glacial acetic acid, rt 1 - 2 hrs
(e) Boc-anhydride, dichloro methane, 2 - 3 hrs
(f) LiOH, THF, MeOH, water, RT
(g) DEPBT, amines or phenylalanine methyl ester, anhydrous THF, triethylamine, RT
(h) Saturated Methanolic KOH in THF (only for 13a)/ 4M HCl in dioxane (for all).
Scheme 1: Synthetic pathway of target compounds 8-13.
Assay for PP-IV inhibition
To determine the DPP-IV inhibition activity of new compounds, we followed previously established standard protocol described in the literature[11-13]. Sitagliptin IC50 was determined as positive control to validate the assay validation. The IC50 values of the new compounds and Sitagliptin are listed in (table 1).
Table 1: Minimized energy of poses and inhibitory activities of docked compounds to DPP-IV active site.
| Ligand | Average (Kcal/mol) | IC50 (ìM) ± SD |
|---|---|---|
| 8 | -8.7 | 1.09 ± 0.070 |
| 9 | -8.7 | 1.35 ± 0.188 |
| 10 | -8.5 | 0.862 ± 0.032 |
| 11 | -8.4 | 0.882 ± 0.068 |
| 12 | -8.2 | 0.876 ± 0.147 |
| 13 | -8.5 | 0.292 ± 0.023 |
| Sitagliptin | -8.4 | 0.023 ± 0.001 |
Material and Methods
Experimental for synthesis
emsp; Reagents and solvents were generally used as received from the commercial suppliers. Tetrahydrofuran (THF) was distilled using sodium and benzophenone in the still under argon. All the reactions for the synthesis of amides were run under anhydrous condition using nitrogen balloon. Melting Points were determined using a MEL-TEMP apparatus and are uncorrected. 1H and 13C NMR spectra were recorded at 90 MHz on an Anasazi instrument Pulse Probe using tetramethylsilane (TMS) as an internal Standard; the values of chemical shift (o) are given in parts per million (ppm) and coupling constants (J) in hertz (Hz). IR spectra were recorded on a Nicolet 10 FT spectrometer with samples placed directly in the path of the light beam. LC/MS (Mass Spectrum) was used to characterize the final test compounds; the value of m/z was reported as M+H peak. Progress of the reaction was monitored by TLC on silica gel plate (Whatman; PE SIL GIUV). Extracts were dried over calcium chloride or magnesium sulfate, and solvent was removed under reduced pressure. Yields refer to the unpurified products which were further purified by recrystallization technique. Aldrich Silica Gel, 70 - 230 mesh, 60A0 was used for column chromatography. Compounds 1 - 7 have been previously reported as referenced under experimental (references (41-48), and were characterized by melting points and spectral data. Intermediate (8a-13a) are new compounds and were characterized by spectral data and melting points, and were used directly to prepare the desired target products (8-13) that were fully characterized by melting points, spectral data and elemental analyses. Elemental analyses were performed by Galbraith Laboratories Inc, Knoxville TN.
Synthesis
5-(1-hydroxy-2-(2, 4, 5-triflourophenyl) ethylidiene)-2,2-dimethyl- 1,3-dioxane-4,6-dione (2)
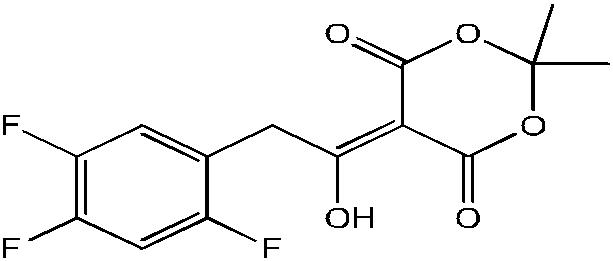
To a 250 ml three necked round bottom flask was added 2, 4, 5-triflourophenyl acetic acid (0.0535 moles), 2, 2-dimethyl- 1, 3-dioxane-4, 6-dione (Meldrum’s acid) (0.0592 moles), N, N-diisopropylethylamine (0.113 moles) and N, N-diimethylamino pyridine (0.0044 moles) in acetonitrile at room temperature. Trimethyl acetyl chloride (0.0580 moles) was added drop wise over 30 minutes to 2 hours while maintaining temperature below 50°C. The reaction was stirred for 3 hours below 55°C and then cooled to 0°C, 1 N HCL was added drop-wise with constant stirring over 30 min to get a precipitate that was filtered to get the crude product (0.0546) of 2 with m.p - 100 - 104, which was dried and recrystallized to obtain pure product (0.0514) 96% yield, m.p – 114-115°C.
1H NMR (CDCl3) δ 1.76 (s, 6H), 4.46 (s, 2H), 7.07 (m, 2H)
13C NMR (CDCl3) δ 27.01, 34.55, 92.08, 104.81, 105.64, 106.80, 119.00, 119.92, 192.83
IR cm-1 3066, 2999, 1731, 1650, 1575, 1523, 1461, 1420, 1382, 1337, 1305, 1263, 1235, 1212, 1176, 1151,1099, 1040, 1023, 956, 919, 893, 852, 844, 788, 744, 703, 659, 644, 620, 569, 546.
3-oxo-4-(2,4,5 triflourophenyl) butanoate (3)
In a 250 ml round bottom flask was added 5-(1-hydroxy-2-(2,4,5-triflourophenyl) ethylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (0.04991 moles) dissolved in toluene/ Methanol(4:1) and set to reflux. Reaction mixture was monitored through TLC (90% EtOAc/hexane). After 4 hrs the reaction was completed as indicated by TLC spot with Rf value-0.4 compared to starting material Rf value-0.6. The reaction mixture was evaporated under vacuum at the end of reaction to get a crude product (oil), which was further washed with brine/EtOAc to get pure product (0..0377 moles) 75.6% yield.
1H NMR (CDCl3) δ 3.55 (s, 2H), 3.74 (s, 3H), 3.85 (s, 2H), 26.98 (m, 2H)
13C NMR (CDCl3) δ 42.20, 48.57, 52.65, 104.62, 105.88, 106.81, 119.27, 119.96, 167.57, 198.56
IR cm-1 2956, 1727, 1634, 1574, 1520, 1434, 1382, 1331, 1235, 1212, 1152, 1099, 1062, 1016, 957, 917, 884, 787, 732, 697, 662, 636, 604, 570.
3-amino-4-(2,4,5,-triflourophenyl) but-2-enoate (4)
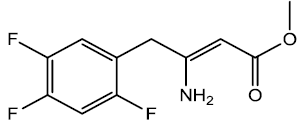
In a 200 ml Round bottom flask was dissolved 3-oxo-4-(2, 4, 5-triflourophenyl) butanoate (0.0377 moles) in 65 ml of methanol. In a separate beaker was dissolved in (0.169 moles) of ammonium acetate in 60 ml of methanol. Transfer the ammonium acetate solution in round bottom flask containing starting material and set to Reflux for 7 - 8 hrs. The reaction was then evaporated to get a crude oil, which was then extracted with weakly alkaline water (pH- 8) and chloroform (2 x 25 ml) to get rid of excess acetic acid. The organic layer then was dried using calcium chloride, filtered and evaporated to get product (0.0359) 95% yield, m.p- 65 - 66°C. The crude product was recrystallized using hexane to get fine needle shape crystals 10.56 g m.p- 70 - 71°C.
1H NMR (CDCl3) δ 3.42 (s, 2H), 3.64 (s, 3H), 4.57 (s, 1H), 6.99(m, 2H)
13C NMR (CDCl3) δ 34.81, 50.46, 85.02, 104.75, 105.67, 105.99, 106.91, 118.13, 119.22, 159.72, 170.56
IR cm-1 3488, 3338, 3085, 3044, 2970, 1663, 1615, 1557, 1505, 1459, 1442, 1422, 1333, 1311, 1264, 1232, 1210, 1194, 1164, 1149, 1085, 1021, 954, 911, 888, 851, 840, 792, 771, 751, 740, 721, 686, 660, 625, 597, 532.
Methyl 3-amino-4-(2,4,5-triflourophenyl) butanoate (5)
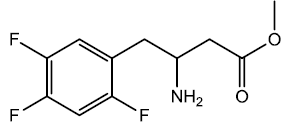
In a 250 ml was added in portions sodium borohydride (0.0788 moles) in 70 ml of acetic acid for about 30 min at 15 - 20°C in Room temperature water bath. The solution was kept stirring until all H2 evolution ceased, after which in a single portion was added enamine (0.0263 moles) to the solution and the reaction mixture was stirred for an hour. Reaction was monitored with TLC at an interval of 30 min in (20% EtOAc/Hexane). The reaction was completed after an hour as indicated by TLC spot at an Rf value 0 compared with sm Rf- 0.4, indicating the amine sticking at the base line. Acetic acid was evaporated under vacuum below 50°C. The residue obtained after evaporation was dissolved in methylene chloride and washed with saturated Sodium carbonate solution (4 x 25 ml). The organic layer was then dried using anhydrous calcium chloride. The organic layer was then evaporated to give β-amino ester (oil) 2.2 g (0.00890 moles) 87.32 % yield.
1H NMR (CDCl3) δ 1.53 (s, 2H), 2.43 (dd, 2H, J = 4, 8 Hz), 2.69 (dd, 2H, J = 3, 5 Hz), 3.44 (m, 1H), 3.69 (s, 3H), 6.97 (m, 2H)
13C NMR (CDCl3) δ 36.46, 41.45, 48.88, 51.77, 104.59, 105.48, 106.79, 118.73, 119.82, 121.58, 122.71, 172.61
IR cm-1 2955, 1730, 1632, 1512, 1438, 1423, 1332, 1208, 1150, 1099, 1038, 1000, 839, 758, 714, 688, 663, 608, 542.
Methyl-3-((tert-butoxycarbonyl) amino)-4-(2,4,5-triflourophenyl) butanoate (6)
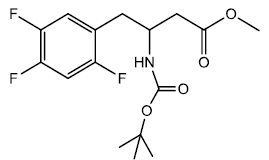
To a stirred solution of amine (0.0220 moles) in 30 ml of dichloromethane was added Boc anhydride (0.0230 moles) dissolved in 30 ml of dichloromethane. The reaction mixture was stirred and left overnight at room temperature. TLC was spotted and found to completion. The reaction mixture was diluted with brine and washed. The organic layer was separated and dried using Calcium chloride. The organic layer was evaporated to get a crude product, m.p- 85 - 92°C. Column chromatography was performed to purify the product to get (0.0190 moles) 86.47% yield, m.p. 97 - 100°C. The crude product was washed with hot petroleum ether to get pure product of 2.500 g (0.00720 moles) m.p- 110 - 111°C.
1H NMR (CDCl3) δ 1.38 (s, 9H), 2.56 (d, 2H, J = 6Hz), 2.86 (d, 2H, J = 7Hz), 3, 70 (s, 3H), 4.14 (m, 1H), 5.14 (d, 1H, J = 9 Hz), 6.99 (m, 2H)
13C NMR (CDCl3) Δ 28.48, 33.32, 38.19, 48.17, 51.98, 79.88, 104.50, 105.77, 106.71, 118.90, 119.63, 155.30, 172.07
IR cm-1 3377, 3058, 2967, 2932, 1735, 1684, 1635, 1515, 1447, 1436, 1421, 1386, 1366, 1355, 1339, 1302, 1266, 1233, 1204, 1153, 1095, 1054, 1035, 996, 914, 897, 886, 852, 840, 777, 758, 727, 689, 666, 594.
Methyl-3-((tert-butoxycarbonyl) amino)-4-(2,4,5-triflourophenyl) butanoic acid (7)
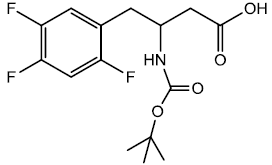
To a solution of β-amino ester (0.00143 moles) in THF/Methanol (10 ml/7.5 ml) was added LiOH•H2O (0.00288 moles) dissolved in 5 ml of H2O. Reaction mixture was stirred at room temperature for 2 hrs monitored continuously with TLC. Reaction mixture was diluted with water after 2 hrs and acidified the pH of reaction mixture between 3 - 4 with 1N HCl. The reaction mixture was then washed with Dichloromethane/Brine, organic layer was dried over anhydrous calcium chloride and evaporated to get Boc protected β-amino acid (0.00119 moles) 83.2% yield; m.p- 143 - 144°C.
1H NMR (CD3COCD3) δ 1.30 (s, 9H), 2.59 (d, 2H, J = 6 Hz), 2.86 (d, 2H, J = 8 Hz), 4.23 (m, 1H), 6.00 (d, 1H, J = 8 Hz), 7.21 (m, 2H)
13C NMR (CDSub>3COCDSub>3) δ 28.41, 29.00, 33.87, 39.39, 48.77, 78.89, 104.79, 106.17, 107.17, 120.01, 120.61, 156.00, 172.80
IR cm-1 3330, 3068, 2983, 1707, 1676, 1534, 1516, 1490, 1451, 1433, 1416, 1392, 1367, 1335, 1308, 1288, 1255, 1233, 1194, 1161, 1093, 1082, 1064, 1039, 1027, 956, 908, 869, 850, 779, 755, 745, 699, 672, 643, 538
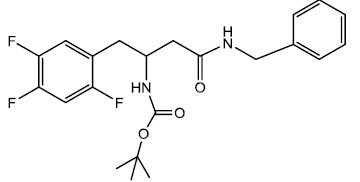
Tert-butyl 4-oxo-4-(phenethylamino)-1-(2,4,5-trifluorophenyl) butan-2-ylcarbamate (8a)
In a 25 ml Round Bottom Flask (r.b.f) 1 was taken 0.300 g (0.000846 moles) of Boc protected β-amino acid and 0.276 g (0.00092) of DEPBT [3-(Diethoxyphosphoryloxy)-(1,2,3)-benzotriazin- 4(3H)-one] in 10 ml of anhydrous THF. To this solution was added initially in equivalent molar quantity 0.092 g (0.00092) of triethylamine and the pH of the solution was adjusted to 8 ~ 9 by adding triethylamine. The solution was set to stir for 30 minutes under inert condition using nitrogen gas. In another round bottom flask was taken amine (phenyl ethylamine) 0.1024 g (0.000846 moles) and dissolved in 5 ml of THF. The amine solution was transferred under anhydrous condition to the Rbf 1 using the dry cannula. The amine containing r.b.f was rinsed with another 5 ml of anhydrous THF and transferred. The reaction was set to stir for 2 hrs. A little white solid started appearing after an hour. After 2 hrs reaction was completed as indicated by TLC (90% CHCl3/ MeOH). The reaction solution was evaporated after and oily residue was obtained. The oily residue obtained was dissolved in EtOAc and was successively washed with 1N HCl (cold 2 x 20 ml), Sat NaHCO3 (2 x 20 ml) and Brine (2 x 20 ml). The organic layer was dried using sodium sulfate and evaporated to give 0.220 g (0.000522 mol) of white solid product. Yield 61.76 % m.p – 182 - 185°C. The white solid product was recrystallized using acetonitrile gave a pure compound with sharp melting point of 185 - 187°C.
1H NMR (DMSO) δ 1.27 (s, 9H), 2.27 (d, 2H, J= 7 Hz), 2.70 (m, 4H overlapping of triplet & doublet J = 7, 7 Hz), 3.25 (m, 3H), 4.00 (m, 1H), 6.70 (d, 1H J = 14 Hz), 7.23 (s, 5H), 7.39 (m, 2H), 7.97 (t, 1H, J= 5, 5 Hz)
13C NMR (DMSO) δ 28.15, 35.33, 47.92, 77.68, 100.43, 126.20, 128.72, 139.65, 169.88
IR cm-1 3340, 2976, 2939, 1682, 1647, 1544, 1523, 1456, 1445, 1423, 1393, 1367, 1353, 1305, 1275, 1231, 1201, 1155, 1092, 1057, 1030, 904, 884, 843, 779, 750, 724, 699, 661, 614, 593, 549.
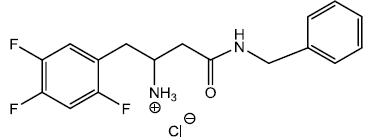
4-oxo-4-(phenethylamino)-1-(2,4,5-trifluorophenyl)butan-2-aminium chloride (8)
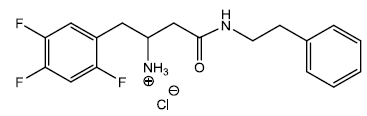
In a dry 5 ml round bottom flask was added 2 ml of 4M HCl/Dioxane solution and stirred in an ice bath for 30 minutes under inert atmosphere. To this ice cold solution was added compound (8) 0.200 g (0.000459 moles) and stirred under inert atmosphere. After 30 min a white precipitate started appearing into the solution. The solution was kept for another 30 min and monitored with TLC (90 % etlylacetate/hexane). The reaction was completed within an hour and the reaction solution was filtered to collect the precipitate 0.101 g (0.000269 mol) yield 58.57 %. The precipitate was washed with diethyl ether to get rid of excess of dioxane and dried. m.p -164 - 166°C. The 40 mg HCl salt (8) was further recrystallize using MeOH/Diethyl ether to get 25 mg of pure salt after 24hrs m.p- 167 - 168°C which was used for elemental analysis and biological testing.
1H NMR (DMSO) δ 2.53, 2.75 (s, d, 3H, overlapping of doublet and singlet, J= 8, 4 Hz), 3.00 (t, 1H, J= 7, 5 Hz), 3.22 (t, 2H, J= 6, 7 Hz), 3.72 (m, 1H), 7.24 (s, 5H), 7.54 (m, 2H), 8.46 (s, 3H)
13C NMR (DMSO) δ 30.92, 35.07, 48.04, 118.73, 119.95, 120.73, 126.27, 128.48, 128.73, 139.49, 168.94 LC/MS (M+H = 337)
IR cm-1 3253, 2923, 2872, 2636, 2574, 2517, 1647, 1604, 1568, 1522, 1455, 1446, 1428, 1383, 1359, 1335, 1305, 1269, 1227, 1211, 1198, 1179, 1154, 1122, 1104, 1058, 1036, 1000, 973, 941, 896, 860, 848, 835, 756, 724, 705, 690, 661, 627, 570, 532. Anal. Calcd for C18H20ClF3N2O: C, 57.99; H, 5.41; N, 7.51. Found: C, 57.29; H, 5.09; N, 7.43
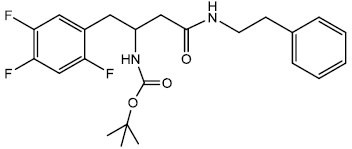
Tert-butyl 4-(benzylamino)-4-oxo-1-(2,4,5-trifluorophenyl) butan-2-ylcarbamate (9a)
In a 25 ml round bottom flask, 1 was taken 0.396 g (0.00112 moles) of Boc protected β-amino acid and 0.389 g (0.0013) of DEPBT [3-(Diethoxyphosphoryloxy)-(1,2,3)-benzotriazin- 4(3H)-one] in 7 ml of anhydrous THF. To this solution was added initially in equivalent molar quantity 0.1315 g (0.0013) of triethylamine and the pH of the solution was adjusted to 8 ~ 9 by adding triethylamine. The solution was set to stir for 30 minutes under inert condition using nitrogen gas. In another round bottom flask was taken amine (Benzylamine) 0.0683 g (0.000563 moles) and dissolved in 5 ml of THF. The amine solution was transferred under anhydrous condition to the Rbf 1 using the dry canula. The amine containing r.b.f was rinsed with another 5 ml of anhydrous THF and transferred. The reaction was set to stir for 2 hrs. After 2 hrs reaction was completed as indicated by TLC (90% CHCl3/ MeOH). The reaction solution was evaporated after and oily residue was obtained. The oily residue obtained was dissolved in EtOAc and was successively washed with 1N HCl (cold 2 x 20 ml), Sat NaHCO3 (2 x 20 ml) and Brine (2 x 20 ml). The organic layer was dried using sodium sulfate and evaporated to give 0.326 g (0.00077 mol) of white solid product. Yield 68.97 % m.p – 163 - 165°C. The white solid product was recrystallized using 1-pentanol gave a pure compound with sharp melting point of 163 - 164°C.
1H NMR (DMSO) δ 1.35 (s, 9H), 2.45 (d, 2H, J= 6 Hz), 2.85 (d, 2H, J= 7 Hz), 4.05 (m, 1H), 4.40 (d, 2H, J= 6 Hz), 5.52 (d, 2H, J= 9 Hz), 6.35 (t, 1H, J= 5, 6 Hz), 6.89 (m, 2H), 7.26 (s, 1H).
IR cm-1 3340, 3065, 2984, 2936, 1686, 1645, 1522, 1455, 1424, 1392, 1367, 1352, 1327, 1305, 1275, 1251, 1232, 1207, 1162, 1151, 1100, 1079, 1057, 1028, 903, 881, 854, 840, 776, 746, 715, 696, 657, 626, 548.
4-(benzylamino)-4-oxo-1-(2,4,5-trifluorophenyl)butan- 2-aminium chloride (9)
In a dry 5 ml round bottom flask was added 2 ml of 4M HCl/Dioxane solution and stirred in an ice bath for 30 minutes under inert atmosphere. To this ice cold solution was added compound (9a) 0.200 g (0.000474 moles) and stirred under inert atmosphere. After 30 min a white precipitate started appearing into the solution. The solution was kept for another 30 min and monitored with TLC (90% EtOAc/hexane). The reaction was completed within an hour and the reaction solution was filtered to collect the precipitate 0.112 g (0.000313 mol) yield 66.00%. The precipitate was washed with diethyl ether to get rid of excess of dioxane and dried m.p – 197 - 200°C. The 40 mg HCl salt (9) was further recrystallize using MeOH/Diethyl ether to get 25 mg of pure after 24hrs salt m.p – 202 - 203°C which was used for elemental analysis and biological testing.
1H NMR (DMSO) δ 2.61 (d, 2H, J= 6 Hz), 3.03 (t, 2H, J= 6, 7 Hz), 3.39 (m, 3H), 3.74 (m, 1H), 4.29 (d, 2H, J= 6 Hz), 7.28 (s, 5H), 7.42 (m, 2H), 8.38 (s, 3H), 8.70 (t, 1H, J= 4, 5 Hz)
13C NMR(DMSO) δ 30.80, 36.69, 42.30, 48.00, 104.95, 106.15, 107.30, 119.77, 120.84, 126.98, 127.50, 128.39, 139.15, 169.06. LC/MS (M+H = 323)
IR cm-1 3270, 3185, 3070, 2924, 2787, 1652, 1607, 1564, 1516, 1498, 1453, 1435, 1424, 1396, 1362, 1330, 1313, 1296, 1274, 1254, 1230, 1214, 1188, 1152, 1137, 1098, 1087, 1055, 1026, 964, 944, 887, 879, 841, 756, 742, 715, 700, 680, 659, 637, 618, 606, 576, 541. Anal. Calcd for C17H18ClF3N2O: C, 56.91; H, 5.06; N, 7.81. Found: C, 56.50; H, 4.84; N, 7.82
Tert-butyl 4-(2,4-dimethoxybenzylamino)-4-oxo-1-(2,4,5-trifluorophenyl) butan-2-ylcarbamate (10a)
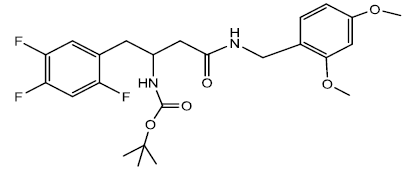
In a 25 ml Round Bottom Flask (r.b.f) 1 was taken 0.500 g (0.00141 mol) of Boc protected β-amino acid and 0.509 g (0.0017) of DEPBT [3-(Diethoxyphosphoryloxy)-(1,2,3)-benzotriazin- 4(3H)-one] in 7 ml of anhydrous THF. To this solution was added initially in equivalent molar quantity 0.192 g (0.0019) of triethylamine and the pH of the solution was adjusted to 8 ~ 9 by adding triethylamine. The solution was set to stir for 30 minutes under inert condition using nitrogen gas. In another round bottom flask was taken amine (2, 4 dimethoxy benzylamine) 0.235 g (0.00141 mol) and dissolved in 5 ml of THF. The amine solution was transferred under anhydrous condition to the Rbf 1 using the dry canula. The amine containing r.b.f was rinsed with another 5 ml of anhydrous THF and transferred. The reaction was set to stir for 2 hrs. After 2 hrs reaction was completed as indicated by TLC (90% CHCl3/ MeOH). The reaction solution was evaporated after and oily residue was obtained. The oily residue obtained was dissolved in EtOAc and was successively washed with 1N HCl (cold 2 x 20 ml), Sat NaHCO3 (2 x 20 ml) and Brine (2 x 20 ml). The organic layer was dried using sodium sulfate and evaporated to give 0.609 g (0.00126 moles) of yellowish white solid product. Yield 89.61 % m.p – 155 - 159°C. The white solid product was recrystallized using acetonitrile gave a pure compound with sharp melting point of 165.5 - 166.5°C.
1H NMR (CDCl3/DMSO) δ 1.35 (s, 9H), 2.83 (d, 2H, J= 7 Hz), 3.79 (s, 3H), 3.82 (s, 3H), 3.99 (m, 1H), 4.35 (d, 2H, J= 6 Hz), 5.68 (d, 1H, J = 9 Hz), 6.16 (t, 1H, J= 5, 5 Hz), 6.43 (d, 2H, J= 7 Hz), 6.97 (m, 2H)
13C NMR (CDCl3/DMSO) δ 28.19, 37.31, 38.83, 41.70, 55.11, 80.06, 98.10, 104.08, 119.02, 129.12, 154.94, 157.94, 159.95, 170.17
IR cm-1 3329, 3060, 2938, 2835, 1679, 1646, 1616, 1590, 1522, 1456, 1421, 1390, 1366, 1354, 1330, 1306, 1277, 1259, 1234, 1204, 1156, 1125, 1095, 1058, 1035, 922, 908, 893, 855, 842, 832, 818, 779, 759, 750, 722, 687, 637, 590, 550.
4-(2,4-dimethoxybenzylamino)-4-oxo-1-(2,4,5-trifluorophenyl) butan-2-aminium chloride (10)
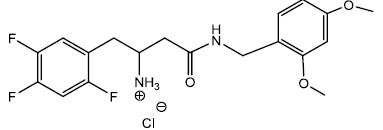
In a dry 5 ml round bottom flask was added 2 ml of 4M HCl/Dioxane solution and stirred in an ice bath for 30 minutes under inert atmosphere. To this ice cold solution was added compound (10a) 0.300 g (0.000622 moles) and stirred under inert atmosphere. After 30 min a white precipitate started appearing into the solution. The solution was kept for another 30 min and monitored with TLC (90% EtOAc/hexane). The reaction was completed within an hour and the reaction solution was filtered to collect the precipitate 0.200 g (0.000478 moles) yield 76.92%. The precipitate was washed with diethyl ether to get rid of excess of dioxane and dried m.p – 154 - 160°C. The 60 mg HCl salt (10) was further recrystallize using MeOH/Diethyl ether to get 40 mg of pure salt after 6 hrs m.p – 163 - 165.5°C which was used for elemental analysis and biological testing.
1H NMR (DMSO) δ 2.57 (d, 2H, J= 6 Hz), 3.01 (t, 2H, J= 6, 5 Hz), 3.75 (s, 3H), 3.77 (m, 1H), 3.79 (s, 3H), 4.14 (d, 2H, J= 5 Hz), 6.44 (m, 2H), 7.08 (d, 2H, J= 8 Hz), 7.57 (m, 2H), 8.42 (s, 3H)
13C NMR (DMSO) δ 30.77, 37.07, 47.92, 55.27, 66.44, 98.26, 104.40, 118.52, 129.23, 157.79, 160.01, 168.96 LC/MS (M+H = 383)
IR cm-1 3344, 3069, 3041, 2749, 2699, 2486, 1638, 1615, 1589, 1516, 1463, 1423, 1388, 1330, 1293, 1254, 1268, 1206, 1186, 1159, 1133, 1109, 1079, 1041, 1032, 907, 888, 860, 834, 758, 725, 680, 659, 641, 619, 581, 538. Anal. Calcd for C19H22ClF3N2O3: C, 54.48; H, 5.29; N, 6.69. Found: C, 54.28; H, 5.19; N, 6.67
Tert-butyl 4-(3-fluorophenethylamino)-4-oxo-1-(2,4,5-trifluorophenyl) butan-2-ylcarbamate (11a)
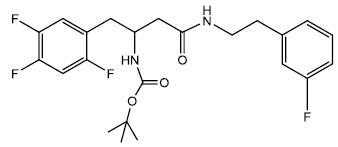
In a 25 ml Round Bottom Flask (r.b.f) 1 was taken 0.500 g (0.00141 moles) of Boc protected β-amino acid and 0.449 g (0.0015) of DEPBT [3-(Diethoxyphosphoryloxy)-(1,2,3)-benzotriazin- 4(3H)-one] in 7 ml of anhydrous THF. To this solution was added initially in equivalent molar quantity 0.152 g (0.0015) of triethylamine and the pH of the solution was adjusted to 8 ~ 9 by adding triethylamine. The solution was set to stir for 30 minutes under inert condition using nitrogen gas. In another round bottom flask was taken amine (3 flouro phenyl ethylamine) 0.196 g (0.00140 moles) and dissolved in 5 ml of THF. The amine solution was transferred under anhydrous condition to the Rbf 1 using the dry canula. The amine containing r.b.f was rinsed with another 5 ml of anhydrous THF and transferred. The reaction was set to stir for 2 hrs. After 2 hrs reaction was completed as indicated by TLC (90% CHCl3/ MeOH). The reaction solution was evaporated after and oily residue was obtained. The oily residue obtained was dissolved in EtOAc and was successively washed with 1N HCl (cold 2 x 20 ml), Sat NaHCO3 (2 x 20 ml) and Brine (2 x 20 ml). The organic layer was dried using sodium sulfate and evaporated to give 0.550 g (0.00121 moles) of yellowish white solid product. Yield 85.91%, m.p – 177 - 182°C. The white solid product was recrystallized using ethyl acetate gave a pure compound with sharp melting point of 182.5 - 183° C.
1H NMR (DMSO) δ 1.28 (s, 9H), 2.33 (d, 2H, J= 7 Hz), 2.73 (m, 4H), 4.03 (m, 1H), 6.70 (d, 1H, J= 9 Hz), 7.05 (d, 3H, J = 9 Hz), 7.30 (m, 3H), 7.97 (t, 1H, J= 5, 4 Hz)
13C NMR (DMSO) δ 28.38, 33.21, 35.15, 42.51, 48.02, 77.92, 112.73, 113.66, 115.23, 116.16, 125.22, 130.40, 130.70, 143.12, 155.17, 157.18, 170.12.
IR cm-1 3341, 2939, 1682, 1645, 1588, 1522, 1489, 1456, 1423, 1393, 1368, 1353, 1305, 1274, 1251, 1231, 1203, 1154, 1093, 1056, 1030, 949, 903, 882, 842, 780, 749, 723, 692, 660, 624, 547.
4-(3-fluorophenethylamino)-4-oxo-1-(2,4,5-trifluorophenyl) butan-2-aminium chloride (11)
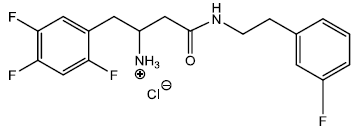
In a dry 5 ml round bottom flask was added 2 ml of 4M HCl/Dioxane solution and stirred in an ice bath for 30 minutes under inert atmosphere. To this ice cold solution was added compound (11) 0.200 g (0.000441 moles) and stirred under inert atmosphere. After 30 min a white precipitate started appearing into the solution. The solution was kept for another 30 min and monitored with TLC (90 % EtOAc/hexane). The reaction was completed within an hour and the reaction solution was filtered to collect the precipitate 0.120 g (0.000308 moles) yield 69.77%. The precipitate was washed with diethyl ether to get rid of excess dioxane, and dried, m.p – 147 - 152°C. The 50 mg HCl salt (10) was further recrystallize using MeOH/Diethyl ether and after 3 days the salt came out of the solution to get 34 mg of pure salt m.p – 152 - 153°C which was used for elemental analysis and biological testing.
1H NMR (DMSO) δ 2.56 (s, 2H, overlapping of peaks in region), 2.63 (m, 2H, Overlap of peaks), 2.97 (m, 2H, overlap of peaks), 3.30 (m, 2H), 3.72 (m, 1H), 7.07 (d, 2H, J= 9 Hz), 7.38 (m, 2H), 7.75 (s, 2H), 8.47 (s, 2 H)
13C NMR (DMSO) 30.73, 34.50, 36.62, 42.29, 47.93, 112.46, 113.37, 114.86, 115.78, 119.55, 120.13, 124.84, 129.95, 130.35, 142.19, 142.51, 168.90
IR cm-1 3354, 3065, 2834, 2758, 2576, 1642, 1586, 1543, 1522, 1508, 1487, 1445, 1427, 1381, 1334, 1312, 1269, 1256, 1244, 1223, 1208, 1176, 1145, 1112, 1095, 1048, 996, 978, 888, 868, 844, 837, 796, 753, 713, 697, 671, 660, 617, 542, 532. Anal. Calcd for C18H19ClF4N2O: C, 55.32; H, 4.90; N, 7.17. Found: C, 54.77; H, 4.63; N, 7.11
Tert-butyl 4-(3,5-dimethoxyphenethylamino)-4-oxo-1- (2,4,5 -trifluorophenyl)butan-2-ylcarbamate (12a)
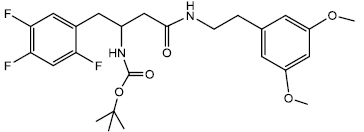
In a 25 ml Round Bottom Flask (r.b.f) 1 was taken 0.500 g (0.00141 moles) of Boc protected β-amino acid and 0.509 g (0.0017) of DEPBT [3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin- 4(3H)-one] in 7 ml of anhydrous THF. To this solution was added initially in equivalent molar quantity 0.243 g (0.0017) of triethylamine and the pH of the solution was adjusted to 8 ~ 9 by adding triethylamine. The solution was set to stir for 30 minutes under inert condition using nitrogen gas. In another round bottom flask was taken amine (3,5 dimethoxy phenyl ethylamine) 0.255 g (0.00141 moles) and dissolved in 5 ml of THF. The amine solution was transferred under anhydrous condition to the Rbf 1 using the dry canula. The amine containing r.b.f was rinsed with another 5 ml of anhydrous THF and transferred. The reaction was set to stir for 2 hrs. After 2 hrs reaction was completed as indicated by TLC (90% CHCl3/ MeOH). The reaction solution was evaporated after and oily residue was obtained. The oily residue obtained was dissolved in EtOAc and was successively washed with 1N HCl (cold 2 x 20 ml), Sat NaHCO3 (2 x 20 ml) and Brine (2 x 20 ml). The organic layer was dried using sodium sulfate and evaporated to give 0.660 g (0.00133 moles) of yellowish white solid product. Yield 94.37% m.p – 147 - 151°C. The yellowish white solid product was recrystallized using acetonitrile gave a pure compound with sharp melting point of 160 - 162°C.
1H NMR (DMSO) δ 1.28 (s, 9H), 2.30 (d, 2H, J= 7 Hz), 2.66 (m, 4H, Overlap of two doublets), 3.35 (m, 3H), 3.72 (s, 6H), 3.92 (m, 1H), 6.26 (d, 3H, J= 4 Hz), 6.71 (d, 1H, J= 8 Hz), 7.32 (m, 2H), 7.96 (t, 1H, J = 5, 5 Hz)
13C NMR (DMSO) δ 28.04, 35.51, 40.52, 47.95, 54.99, 77.59, 98.05, 106.62, 141.82, 154.86, 160.50, 169.76
IR cm-1 3324, 2940, 2838, 1678, 1644, 1615, 1596, 1523, 1470, 1455, 1424, 1392, 1367, 1334, 1306, 1285, 1252, 1232, 1208, 1152, 1104, 1079, 1049, 1017, 993, 921, 889, 853, 839, 824, 780, 757, 696, 660, 622, 608, 563, 538.
4-(3,5-dimethoxyphenethylamino)-4-oxo-1-(2,4,5-trifluorophenyl) butan-2-aminium chloride (12)

5 In a dry 5 ml round bottom flask was added 2 ml of 4M HCl/Dioxane solution and stirred in an ice bath for 30 minutes under inert atmosphere. To this ice cold solution was added compound 0.250 g (0.000504 moles) and stirred under inert atmosphere. The solution was kept for an hour and monitored with TLC (90% EtOAc/hexane). The reaction was completed after an hour and the reaction mixture was evaporated. The residue was dissolved in water in a round bottom flask, kept in an ice bath, and the pH of the solution was adjusted to 8 ~ 9 using sodium bicarbonate and the aqueous layer were extracted with ethyl acetate. The organic layer was dried using calcium chloride and evaporated to get 0.120 g (0.000303 moles) yield 60.13%. The precipitate was washed with diethyl ether to get rid of excess of dioxane and dried m.p – 150 - 154°C. The 40 mg HCl salt (12) was further recrystallize using MeOH/Diethyl ether/HCl in dioxane and after 24 hrs the salt came out of the solution to get 24 mg of pure salt m.p –154 - 155°C which was used for elemental analysis and biological testing.
1H NMR (DMSO) δ 2.63 (m, 4H), 2.96 (t, 2H, J = 6, 7 Hz), 3.19 (m, 2H), 3.71 (s, 6H), 3.71 (m, 1H, Overlap of 6H & 1H), 6.45 (s, 3H), 7.47 (m, 2H), 8.45 (m, 4H)
13C NMR (DMSO) δ 30.81, 47.97, 55.11, 98.11, 106.68, 141.73, 159.11, 160.52, 168.91 LC/MS (M+H = 383). Anal. Calcd for C20H24ClF3N2O3: C, 54.49; H, 5.59; N, 6.47. Found: C, 54.43; H, 5.35; N, 6.84
Methyl2-(3-((tert-butoxycarbonyl) amino)-4-(2,4,5-trifluorophenyl) butanamido) -3-phenylpropanoate (13aa, methyl ester of 13i)
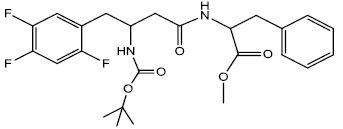
In a 25 ml Round bottom Flask 1 was dissolved Boc protected β-amino acid 2.00 g (0.0056 moles), DEPBT [3-(Diethoxyphosphoryloxy)-(1,2,3)-benzotriazin-4(3H)-one] 1.80 g (0.00080 moles) and triethylamine 0.081 g (0.00080 moles) in anhydrous THF (5 ml). In another 25 ml round bottom flask 2 was dissolved L-Phenylalanine methyl ester 1.004 g (0.00056) which was then transferred in anhydrous condition through a cannula into the flask 1. The pH of the solution was adjusted to 8 ~ 9 by adding triethylamine and the reaction mixture was stirred at room temperature for about 2 hrs. The reaction was followed by TLC (90% CHCl3/ MeOH). At the end of the reaction, solvent was evaporated under vacuum and the oily residue found was dissolved in EtOAc. The organic layer was successively washed with 1N HCl (cold 2 x 20 ml), Sat NaHCO3 (2 x 20 ml) and Brine (2 x 20 ml). The organic layer was dried using sodium sulfate and evaporated to give yellowish white product 2.450 g (0.00494 moles). Yield 88.21 %, m.p- 155 - 157°C. The Crude yellowish white solid was washed with 1-pentanol and hexane to get fine white solid with m.p- 162 - 164°C.
1H NMR (d6-DMSO) δ 1.28 (s, 9H), 2.30 - 2.77 (m, 4H, overlapping doublets), 2.99 (d, 2H, J = 9 Hz), 3.62 (s, 4H overlapping of methyl with methine proton), 4.01 (m, 1H), 4.55 (m, 1H), 6.68 (d, 1H, J = 9 Hz), 6.99 (m, 2H), 7.25 (s, 5H), 8.45 (d, 2H, J = 8 Hz)
13C NMR (d6-DMSO) δ 28.17, 32.83, 36.91, 47.83, 51.93, 53.59, 77.74, 105.60, 106.43, 119.18, 119.94, 122.61, 126.64, 128.39, 129.20, 137.41, 154.94, 170.11, 172.17
IR cm-1 3338, 2978, 1741, 1683, 1646, 1520, 1445, 1423, 1391, 1367, 1354, 1336, 1275, 1253, 1216, 1156, 1096, 1051, 1024, 981, 903, 888, 841, 753, 696, 660, 608.
2-(3-(tert-butoxycarbonylamino)-4-(2,4,5-trifluorophenyl) butanamido)-3- phenylpropanoic acid (13a)
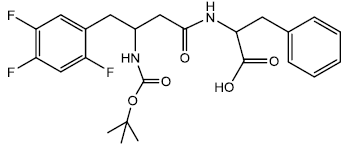
In a 25 ml of round bottom flask was taken 1.00 g (0.00202 moles) of compound 13aa. The compound was dissolved in 12/2 ml of anhydrous THF/Methanol. The solution was stirred for 30 minutes in ice bath. To this cold solution was added 2 ml of 2N Methanolic KOH and the solution was stirred for an hour. The reaction was monitored using TLC (90% EtOAc/hexane). The reaction was completed after 2 hrs and white solid salt precipitating out of the reaction mixture was observed. The reaction mixture was completely evaporated after the completion of reaction to dryness. The dry white solid salt was dissolved in 30 ml of cold distilled water and stirred until all the residue was dissolved, after this the pH of the solution was adjusted to pH 3 ~ 4 using 1N HCl. The precipitate was observed after adjusting the pH which was extracted with diethyl ether/ethyl acetate (4:3, 4 x 25 ml). The organic layer was separated and dried using anhydrous sodium sulfate and evaporated to dryness under vacuum to get 0.891 g (0.00186 moles) of white solid product yield- 92%. m.p - 183.5 - 185.5°C.
1H NMR (DMSO) δ 1.27 (s, 9H0, 2.34 (m, 4H, Overlap of doublet and doublet), 3.00 (dd, 2H, J=10, 6 Hz), 3.99 (m, 1H), 4.51 (m, 1H), 6.66 (d, 1H, J= 9 Hz), 7.24 (s, 5H, 7.48 (m, 2H), 8.28 (d, 1H, J= 8 Hz)
13C NMR (DMSO) δ 28.07, 36.60, 47.74, 53.37, 77.65, 119.08, 119.94, 122.45, 123.36, 126.41, 128.24, 129.16, 137.75, 154.82, 170.01, 173.10
IR cm-1 3336, 2977, 1705, 1679, 1651, 1522, 1446, 1423, 1392, 1367, 1354, 1339, 1279, 1233, 1203, 1153, 1097, 1053, 1030, 906, 886, 843, 779, 752, 722, 696, 666, 611, 564, 542.
4-((1-carboxy-2-phenylethyl) amino)-4-oxo-1-(2,4,5-trifluorophenyl) butan-2-aminium chloride (13)
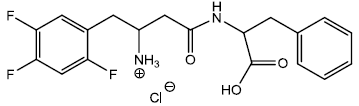
In a dry 5 ml round bottom flask was added 2 ml of 4M HCl/Dioxane solution and stirred in an ice bath for 30 minutes under inert atmosphere. To this ice cold solution was added compound (13a) 0.250 g (0.000520 moles) and stirred under inert atmosphere. The solution was kept for an hour and monitored with TLC (90 % EtOAc/hexane). The reaction was completed after an hour and the reaction mixture was evaporated. The residue was washed with (2 x 25 ml) diethyl ether and evaporated, after an initial wash the residue was again dissolved in diethyl ether and decanted to get rid of impurities. The residue was dried in a vacuum at 40°C for 3 hrs to get 0.120 g (0.000288 moles) yellowish white solid yield 60.13%. The precipitate was washed with diethyl ether to get rid of excess of dioxane and dried m.p– 82 - 89°C. The 40 mg HCl salt (10a) was further recrystallize using MeOH/Diethyl ether/HCl in dioxane and after 24 hrs the salt came out of the solution to get 24 mg of pure salt m.p –112 - 114.5°C which was used for elemental analysis and biological testing.
1H NMR (DMSO) δ 2.52 (s, 2H), 2.94 (m, 3H, Overlap of protons), 3.58 (m, 2H, some residue of dioxane at 3.58 also seen), 4.46 (m, 1H), 7.25 (s, 5H), 7.55 (m, 2H), 8.66 (d, 2H, J= 8 Hz)
13C NMR (DMSO) δ 30.57, 36.86, 47.86, 53.77, 119.48, 120.57, 126.54, 128.29, 129.24, 137.66, 169.29, 172.88
IR cm-1 3228, 3141, 3034, 2930, 2849, 1733, 1713, 1678, 1655, 1615, 1514, 1456, 1427, 1378, 1355, 1334, 1260, 1211, 1174, 1157, 1101, 1061, 1037, 990, 953, 897, 836, 777, 756, 724, 704, 689, 614, 590, 571, 534. LC/MS (M+H = 381). Anal. Calcd for C19H20ClF3N2O3: C, 54.75; H, 4.95; N, 6.72. Found: C, 54.37; H, 5.38; N, 6.28
Results
Molecular Modeling
All compounds were subjected to energy minimization before docking into the crystal structure catalytic site. The energy minimization and docking were accomplished using “Vina” software and “Pyrx’’ (V.08) program which provides a Microsoft Windows interface for Vina[14,15].
Docking results were visualized through discovery studio software[16]. The validity of the docking process was assessed by comparing the docking mode of Sitagliptin-lowest energy pose, generated by Pyrx, with the crystal structure mode of binding as shown in figure 4. The modeling process starts with downloading the crystal structure of sitaglipting docked with DPP-IV in a pdb format (PDB ID 1 x 70) from the protein databank (www.rcsb.org). To prepare the protein for docking studies; the .pdb file was imported to Autodock Tools (v.1.5.6). Water molecules and the ligand (sitagliptin) were removed from the structure, the polar hydrogen’s were added to the residues and the structure was converted to a .pdb qt file format in which the partial charges on the residues were calculated. To prepare the ligands for docking, the structure of the ligands was drawn using Marvin Sketch (V.15.11.9) then saved as a .sdf file format[17]. The protein as well as the ligands files were then imported to Pyrx software where the energy of ligands were minimized using Open Babel extension in the program, then the files converted to .pd bqt format and save[18]. The grid box for the docking was set to be 25 A° X 25 A° X and was centered on the active site which is located at the following coordinates: X = 40.1242 , Y = 50.9588 , Z = 36.1963. The docking studies were performed three times and the potential energies were reported as average of the three runs as summarized in (Table-1). The docking files were then imported to Discovery Studio Visualizer (V. 4.5.0.15071) to visualize the interactions and binding modes of Sitagliptin and the designed compounds. Modeling process was validated by comparing the binding mode of the energy minimized pose of Sitagliptin with that observed in the crystal structure. (Figure 4) depicts that the energy minimized pose of the drug is overlaying on the crystal structure pose confirming the validity of the docking process. (Figure 5) (a and b) depicts the docking modes of all six compounds: alone (figure 5a), and superimposed on sitagliptin crystal structure mode (figure 5b). Figures 5c and 5d are simplified figures of 5a and 5b, respectively, without the enzyme skeleton.
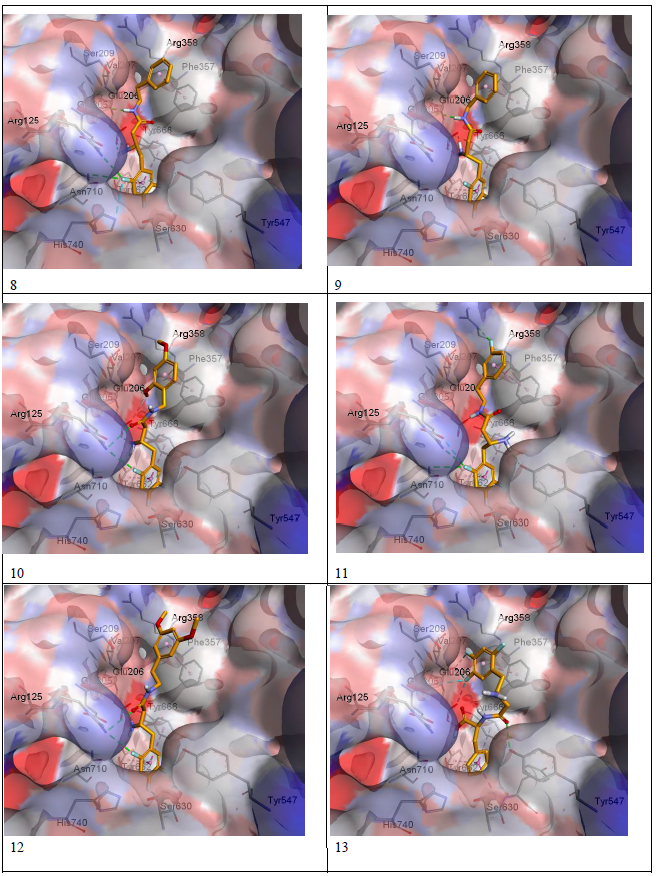
Figure 5a: Designed molecules docked in DPP-IV crystal active site.
Interestingly, the energy minimization studies of the six compounds and Sitagliptin revealed very comparable energy minimum values as listed in (table-1). Those values are reflection of the binding affinities to the active site. (Table-1) data indicate that the designed lipophilic areas of the new compounds resulted in comparable affinities to that of Sitagliptin to the catalytic site. (Figure 5 a-d) with new compounds docked into the active site, also confirms that the lipophilic entities introduced in the new compounds are oriented facing the lipophilic amino acid residue Phe 357 in the S2 pocket. (Figure- 5 a-d), also depicts super imposable binding mode with sitagliptin in the catalytic site. However, although the hydrophobic interaction with Phe 357 were observed, and the compounds are general super imposable on sitagliptin pose, (figure- 5 a-d), the activity of the designed compounds were approximately an order of magnitude less active than sitagliptin, (table-1). The IC50 values of compounds 8 - 12 were generally unaffected by the substitutions on the aromatic ring, nor by the distance the aromatic ring from the β-amino amide. This may signify that other interactions of the triazolo-piperazine ring system of Sitagliptin such as electrostatic and hydrogen bonding may play more important role in the binding process than just the hydrophobic interactions. Another interesting observation from the docking studies; that all designed compounds, except compound 13 which is a carboxylic acid derivative of compound 8, were found to be with almost full superimposition poses over the standard drug in the active site (5a-d). However, interestingly, when compound 13 when docked into the active site it was found to bind in a flipped over mode. The carboxylic group instead of heading toward Arg358 the S-2 pocket, to form ionic interaction, was found to be orient the entire molecule into a flipped over mode with new and different set of interactions with the active site. The trifluro phenyl ring is now facing Phe357, and two of its fluro groups forming new bonds with Glu205, Glu206, Tyr666. The amide carbonyl has interaction with Tyr547 hydroxyl group, and the carboxylic group headed down away from the S2 pocket to interact with Tyr666 hydroxyl group (figure 5c-13). The new set of interactions, possibly, contributes to the 5-fold increase in activity of compound 13 over the rest of the series and to its closer activity to that of Sitagliptin. Further structural modifications are possible to enhance the binding of compound 13, for possible increase in affinity, hence, higher activity.
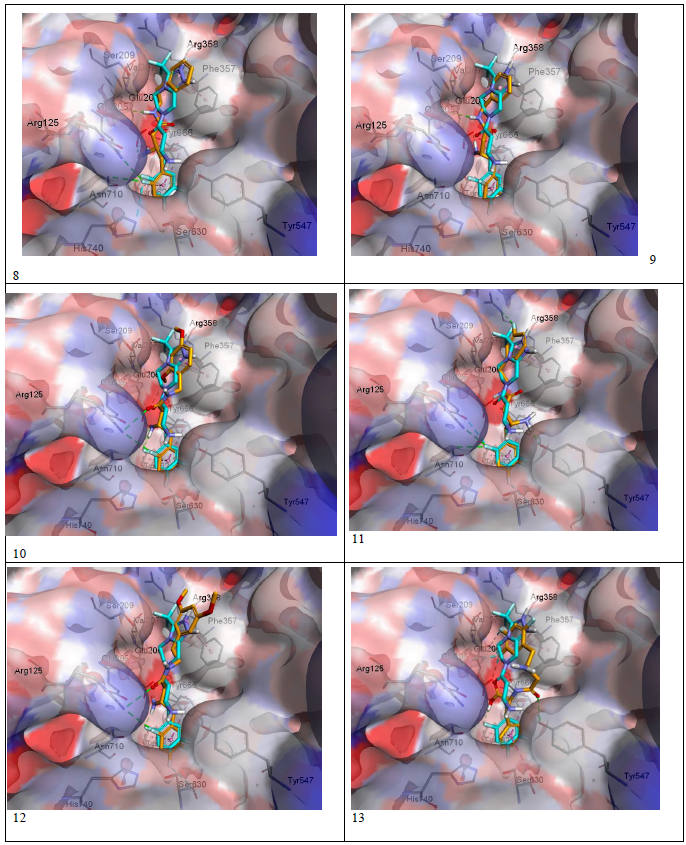
Figure 5b: Figure 5b: Compounds 8-13 (orange) overlaid on sitagliptin (blue).
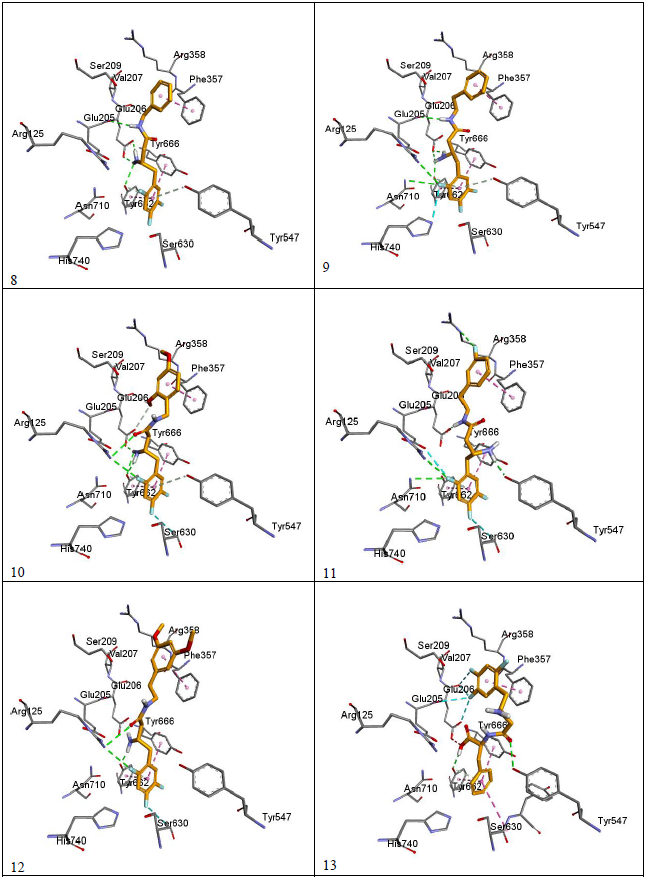
Figure 5c: Simplified Docking for compounds 8-13 with the catalytic site of DPP-IV.
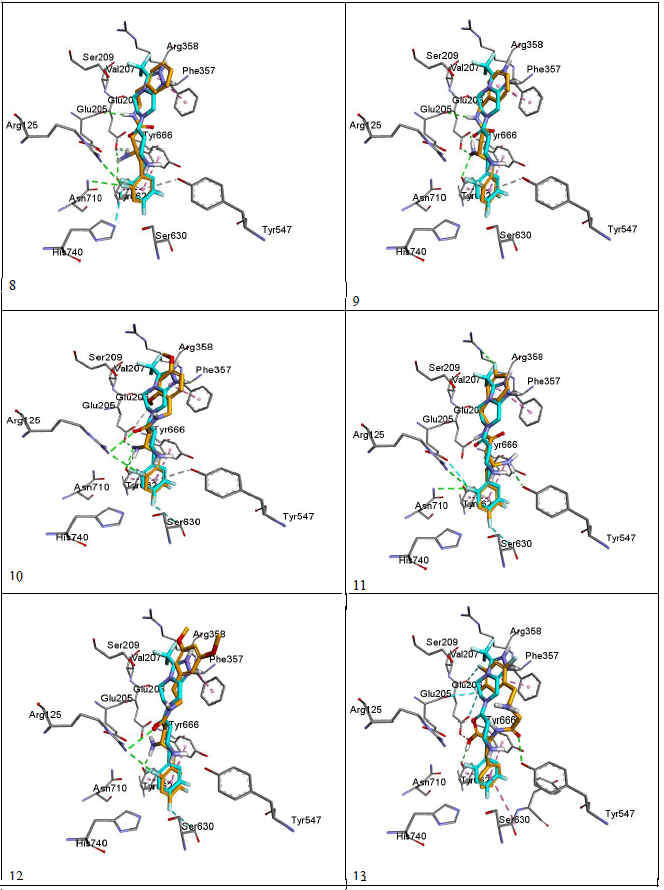
Figure 5d: Overlaying of the compounds (orange) with sitagliptin (blue).
Conclusion
The designed compounds were successfully docked into the catalytic active site of the enzyme with mostly super imposable pose over Sitgliptin crystal structure. This proves that the design by modeling was successful in producing molecules with affinity to the catalytic site. The affinity values of the energy minimized poses were almost equal to that of Sitagliptin, indicating similar interaction modes. Although the new compounds were nearly super imposable on Sitagliptin in the active site, and the affinity values are close, yet the >in vitro activity was not parallel to the observed docking data. Compound 13 showed different flipped over docking mode than that of Sitagliptin and the other five new derivatives. Combining the observed flipped over mode with the observed 5-fold increase in vitro activity over compounds 8 - 12 signifies that a new series of inhibitors are at hand that requires further exploration. This finding may give molecular modeling some value for drug design when the binding poses are considered in addition to the affinity and superimposition factors.
Acknowledge:
The authors are thankful to MCPHS University for funding this research, no grant money was used.
Conflict of interest:
The authors declare that they have no conflict of interest.
References
1. Holst, J.J., Gromada, J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. (2004) Am J Physiol Endocrinol Metab 287(2): El99-E206.
Pubmed ||Crossref|| Others
2. Holz, G. , Epac, A. New cAMP-Binding Protein in Support of Glucagon like Peptide-1 Receptor- Mediated Signal Transduction in the pancreatic β- -Cell. (2004) Diabetes 53: 5- 13.
Pubmed||Crossref||Others
3. Thornberry, N., Weber, A. Discovery of JANUVIA (Sitagliptin), a Selective Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. (2007) Curr Top Med Chem 7(6): 557-568.
Pubmed ||Crossref|| Others
4. Mehanna, A. Anti-diabetic Agents: Past, Present and Future. (2013) Future Med Chem 5(4): 411-430.
Pubmed ||Crossref|| Others
5. Dooseop, K., Kowalchick, J.E., Brockunier, L.L., et al. Discovery of Potent and Selective Dipeptidyl Peptidase IV Inhibitors Derived from β--Aminoamides Bearing Subsituted Triazolopiperazines. (2008) J Med Chem 51(3): 589-602.
Pubmed ||Crossref|| Others
6. Xu, F., Armstrong, J.D 3rd., Zhou, G.X., et al. Mechanistic Evidence for an β-Oxoketene Pathway in the Fonnation of β-Ketoamides/Esters via Meldrum's Acid Adducts. (2004) J Am Chem Soc 126(40):13002-13009.
Pubmed ||Crossref|| Others
7. Hsiao, Y., Rivera, N.R., Rosner, T., et al. Highly Efficient Synthesis of β-Amino Acid Derivatives via Asymmetric Hydrogenation of Unprotected Enamines. (2004) J Am Chem Soc 126(32): 9918-9919.
Pubmed ||Crossref|| Others
8. Ye, Y.H., Li, H., Jiang, X. DEPBT as an Efficient Coupling Reagent for Amide Bond Formation with Remarkable Resistance to Racemization. (2005) Biopolymers 80(2-3): 172-178.
Pubmed ||Crossref|| Others
9. Liu, P., Sun, L.P., Chen, B., et al. Application of DEPBT for synthesis of N-protected peptide alcohols and its derivatives. (2002) Synthetic communications 32(3): 473-480.
Pubmed||Crossref|| Others
10. Han, G., Tamaki, M., Hruby, V.J. Fast, efficient & selective deprotedtion of the tert-butoxy carbonyl (boc) group using HCl/dioxane (4M). (2001) J Pept Res 58(4): 338-341.
Pubmed ||Crossref|| Others
11. Lankas, G.R., Leiting, B., Roy, R.S., et al. Dipeptidyl Peptidase IV Inhibition for the Treatment of Type 2 Diabetes Potential Importance of Selectivity Over Dipeptidyl Peptidases 8 and 9. (2005) Diabetes 54(10): 2988-2994.
Pubmed ||Crossref|| Others
12. Kato, T., Nagatsu, T., Kimura, T., et al. Fluorescence assay of X-prolyl dipeptidyl-aminopeptidase activity with a new fluorogenic substrate. (1978) Biochem Med 19(3): 351-359.
Pubmed ||Crossref|| Others
13. Pryor, L.B., Wu, K., Marsilio, J.F., et.al. Catalytic properties and inhibition of proline-specific dipeptidyl peptidases II, IV and VII. (2003) Biochem J 371: 525-532.
Pubmed ||Crossref|| Others
14. Dallakyan, S., Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. (2015) Methods Mol Biol 1263: 243-250.
Pubmed ||Crossref|| Others
15. Trott, O., Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. (2010) J Comput Chem 31(2):455-461.
Pubmed ||Crossref|| Others
16. Biovia Discovery Studio Visualizer. 2015.
Pubmed||Crossref|| Others
17. https://www.chemaxon.com/products/marvin/marvinsketch/. 2015.
Pubmed||Crossref||Others
18. O’Boyle, N.M., Banck, M., James, C.A., et al. Open Babel: An Open chemical toolbox. (2011) J Cheminform 3(10): 33.
Pubmed ||Crossref|| Others










