
Effect of Morin on Pharmacokinetics of Prasugrel in Rats & In Vitro Metabolic Stability Followed by UPLC Method
Kapendra Sahu1,2* and Anees A. Siddiqui1*
Affiliation
- 1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Jamia Hamdard (Hamdard University), Hamdard Nagar, New Delhi (India)-110062.
- 2National Dope Testing Laboratory, MYAS, Government of India, Lodhi Road, New Delhi (India) -110003
Corresponding Author
Kapendra Sahu., Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Jamia Hamdard (Hamdard University), Hamdard Nagar, New Delhi (India)-110062, Tel: +91-11-26059688/5650; Fax: +91-11 27048685; E-mail: kapendra_sahu@yahoo.com
Citation
Sahu, K., et al. Effect of Morin on Pharmacokinetics of Prasugrel in Rats & In Vitro Metabolic Stability Followed by UPLC Method. (2017) J Med Chem Toxicol 2(2): 79-84.
Copy rights
© 2017 Sahu, K. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Prasugrel; Human Liver Microsomes; Pharmacokinetics; CYP3A; in vivo; in vitro; UPLC
Abstract
The aim of present study was to investigate the effect of Morin on the pharmacokinetics of Prasugrel, a substrate of P-glycoprotein (P-gp) and Cytochrome 3A (CYP3A), in rats, and metabolic stability (high throughput) studies using human liver microsomes in UPLC. For pharmacokinetics studies, Male wistar rats were pretreated with Morin (10 mg/kg) for 1 week and on the last day, a single dose of Prasugrel (1 mg/ kg) was given orally. In another group, both morin and Prasugrel were co-administered to evaluate the acute effect of morin on Prasugrel. The control group received oral distilled water for 1 week and administered with Prasugrel on the last day. As Morin is a known inhibitor of P- Glycoprotein (P-gp) and CYP 3A, it was expected to improve the bioavailability of Prasugrel. Surprisingly, the area under the concentration–time curve and peak plasma concentration relative to control of Prasugrel were 1.50- and 1.45- fold, respectively, greater in the morin-pretreated group. However, co-administration of morin had no significant effect on these parameters. Prasugrel dosages should be accustomed to avoid concomitant for potential drug interaction when Prasugrel is used clinically in combination with morin and morin-containing dietary supplements. Apart from aforementioned merits, the results of this study are further confirmed by clinical trials.
Introduction
Prasugrel is a prodrug of piperazine derivative (Figure 1) that inhibits platelet aggregation in vivo by antagonism of the P2Y12 class of platelet purinergic receptors[1-3]. Subsequent studies showed that the conversion of active metabolite is mediated largely by Cytochrome (CYP) 3A4 and CYP2B6 and, to a lesser extent, by CYP2C9 and CYP2C19[4,5].
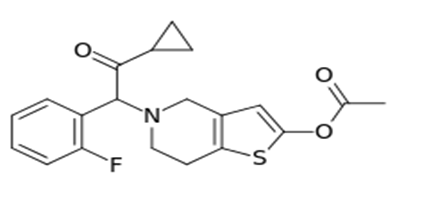
Figure 1: Chemical structure of Prasugrel.
Morin (2-(2,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one) is a flavonoid (flavonol) constituent of many herbs and fruits. In vitro studies have shown morin to have a variety of beneficial activities, including anti-cancer, antioxidation, and anti-inflammation etc[6,7]. Now a day in vitro metabolic stability is incorporated into the drug discovery process. These studies facilitate analytical approach comprised specificity and higher throughput. By understanding the metabolic stability of compounds early in discovery, compounds can be ranked for further studies and the potential for a drug candidate to fail in development as a result of pharmacokinetic reasons may be reduced[7]. The full Pharmacokinetics (PK) studies, rapid screens offer significant savings in the in-life portion by standardizing dosing and study protocols, eliminating study arms, reducing animal usage and limiting the number of sampling points[8,9]. Buening.,et al. also reported that morin could inhibit cytochrome P-450 reductase in human liver microsomes. Orally administered morin, act as P-gp and CYP3A inhibitors, may provide inhibition of platelets aggregation effects to ameliorate the bioavailability of Prasugrel in combination therapy. Therefore, the purpose of this study examined the effect of morin on the bioavailability and pharmacokinetics of Prasugrel in rats. So far to our knowledge there was no method has been reported for UPLC method yet on the method development, effect of morin on pharmacokinetics of Prasugrel, and in vitro metabolic stability studies for this drug.
Experimental
Chemicals
Prasugrel and clopidogrel were obtained as a gratis sample from Micro Labs. Limited (Bangalore, India). Morin, Midozolam and 6-Hydroxy Midozolam were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). HPLC grade methanol and acetonitrile were obtained from the Merck Co. (Darmstadt, Germany). Pooled human liver microsomes from 30 donors were purchased from BD Gentest (Woburn, MA). Ammonium acetate, magnesium chloride and ethyl acetate were all obtained from Merck (Germany). NADPH reduced tetrasodium salt was purchased from Sisco Research Laboratories, (Mumbai, India). Potassium dihydrogen ortho-phosphate, orthophosphoric acid and potassium hydroxide were purchased from Sigma Aldrich Ltd (St Louis, USA). Milli-Q pure water was obtained from a Millipore Elix water purification system purchased from Millipore India Pvt. Ltd. (New Delhi, India). All other chemicals were of analytical grade.
Animals and legal prerequisite
Young, adult male Wistar rats, weighing 200 - 230 g, were procured from the Institutional Animal Ethics Committee (IAEC), Jamia Hamdard (New Delhi, India). Rats were housed in well ventilated cages at room temperature (24 ± 2°C) and 40 - 60 % relative humidity while on a regular 12 h light-dark cycle. The animals were acclimatized for a minimum period of 3 days prior to the experiment. Approval from the Committee was sought and the study protocols were approved before the commencement of the studies.
Instrumentation
Ultra Performance Liquid Chromatography (UPLC) UPLC was performed with a binary solvent delivery pump, an auto sampler and PDA detector of Acquity UPLC system manufactured by Waters Corporation; Milford, Massachusetts, USA; data were acquired and processed using Empower software. The chromatographic separation was performed using a Waters Acquity BEH 10 × 2.1 mm, 1.7 μm, C18 column.
UPLC conditions for plasma sample analysis
Prasugrel was estimated in plasma samples using UPLC method. The chromatographic separation was performed using a Waters Acquity BEH 10 × 2.1 mm, 1.7 μm, C18 column. The mobile phase containing a mixture of acetonitrile and water in the ratio of 30:70 (v/v) at a flow rate of 0.2 mL/min was used. The detection was obtained at a wavelength of 240 nm. The injection volume was 3 μL; mobile phase was used as a diluent while the column was maintained at 30°C. These studies were carried out using a photo diode array detector for checking purity of peaks.
Pharmacokinetics of morin
Wistar Rats were divided into three groups of each of five: the control group (Prasugrel 30 mg/kg, oral), co-administration group (10 mg/kg of morin orally co-administered with 30 mg/kg of Prasugrel) and pretreatment group (1 mg/kg of Prasugrel was administered orally after 1-week pretreatment with morin 10 mg/kg). Blood samples (approx. 150 μl) from retro-orbital plexus were collected into heparinized microfuge tubes at 0.5, 1, 1.5, 2, 4, 8, 12, and 24 h post-dosing and plasma was harvested by centrifuging the blood at 14,000 rpm for 10 min and stored frozen at -20°C until bioanalysis.
Preparation of stock and standard solutions
The primary stock solutions of the analyte (Prasugrel; 5 mg/mL) and IS (Clopidogrel); 200 μg/mL) were prepared in water and stored at 4°C. Appropriate dilutions were made in water for Prasugrel to produce working stock solution of 0.01, 0.02, 0.1, 0.5, 1 & 2 mg/mL and on the day of analysis this set of stocks was used to prepare standards for the calibration curve. Another set of working stock solutions of Prasugrel was made in Triple Distilled Water (TDW) at 0.2, 2, and 15 μg/mL for preparation of QC samples. Individually QC and CC working stock solutions of Prasugrel were spiked into blank plasma for QC and CC samples. Calibration plots were constructed in the range 0.1 – 20 μg/mL for Prasugrel in rat plasma (concentrations 0.1, 0.2, 1, 5, 10 and 20 μg/mL). Calibration standards were prepared by spiking 90 μL of control pooled rat plasma with the appropriate working solution of Prasugrel (10 μL) and IS (10 μL) on the day of analysis. Samples for the determination of precision and accuracy were prepared by spiking control rat plasma in bulk at four concentration levels [40 ng/mL (lower limit of quantitation, LLOQ), 0.2 μg/mL (QC low), 2 μg/mL (QC medium) and 15 μg/mL (QC high)] and 100 L volumes were aliquoted into different tubes and stored at -20°C until analysis.
Sample preparation
A simple liquid–liquid extraction method was followed for extraction of Prasugrel from rat plasma. To 100 μL of plasma in a tube, 10 μL of IS solution (200 μg/mL in water), vortex mixed for 30 seconds on a cyclomixer (Spinix Tarsons, Kolkata, India). Next a 3 mL aliquot of extraction solvent, butanol was added. The mixture was then vortexed for 5 min, followed by centrifugation for 5 min at 3000×g at 25°C on Sigma 3 - 16K (Frankfurt, Germany). The organic layer of 2 ml was separated and evaporated to dryness under vacuum in speedvac concentrator (Savant Instrument, Farmingdale, USA). The residue was reconstituted in 100 μL of the mobile phase and 3 μL was injected into the UPLC system for analysis.
Validation Procedures
A full validation according to the FDA guidelines was performed for the assay in rat plasma.
Recovery
The recovery of Prasugrel and IS, through liquid–liquid extraction procedure, was determined by comparing the responses of the analytes extracted from replicate QC samples (n = 6) with the response of analytes from post-extracted plasma standard (spiked blank plasma extracts after extraction) sample at equivalent concentrations. Recoveries of Prasugrel were determined at QC low, QC medium and QC high concentrations, viz., 0.2, 2, and 15 μg/mL, where as the recovery of the IS was determined at a single concentration of 2 μg/mL.
Specificity and Selectivity
The specificity of the method was evaluated by analyzing rat plasma samples collected from six different rats to investigate the potential interferences at the UPLC peak region for analyte and IS using the proposed extraction procedure for chromatographic conditions.
Matrix Effec
The effect of rat plasma constituents over the separation of Prasugrel and IS was determined by comparing the responses of the post-extracted plasma standard QC samples (n = 6) with the response of analytes from neat standard samples (10 μL of required working stock sample spiked into 90 μL of water instead of blank plasma) at equivalent concentrations[10,11]. The matrix effect for Prasugrel was determined at QC low, QC medium and QC high concentrations, viz., 0.2, 2 and 15 μg/mL whereas the matrix effect over the IS was determined at a single concentration of 200 ng/mL.
Calibration Curve
The calibration curve was developed by plotting the ratio of the peak area of Prasugrel to that of IS versus the nominal concentration of calibration standards. The final concentrations of calibration standards obtained for plotting the calibration curve were 0.1, 0.2, 1, 2, 5, 10 & 20 μg/mL. The response for Prasugrel was strictly linear in the investigated concentration range of a correlation coefficient (r²) of 0.9995. The acceptance criteria for each back-calculated standard concentration were ±15% deviation from the nominal value except at LLOQ, which was set at ± 20%[10,11].
Precision and Accuracy
The intra-day assay precision and accuracy were estimated by analyzing six replicates at four different QC levels, i.e., 0.2, 2 and 15 μg/mL. The inter-assay precision was determined by analyzing the four levels QC samples on three different runs. The criteria for acceptability of the data included accuracy within ± 15% Standard Deviation (S.D.) from the nominal values and a precision of within ± 15% Relative Standard Deviation (R.S.D.), except for LLOQ, where it should not exceed ± 20% of accuracy as well as precision.
Stability Experiments
All stability studies were conducted at two concentration levels, i.e. QC low and QC high, using six replicates at each concentration levels. Replicate injections of processed samples were analyzed up to 18 h to establish auto-sampler stability of analyte and IS at 4°C. The peak areas of analyte and IS obtained at initial cycle were used as the reference to determine the stability at subsequent points.
The stability of Prasugrel in the biomatrix during 4 h exposure at room temperature in rat plasma (bench top) was determined at ambient temperature (25 ± 2°C). Freeze/thaw stability was evaluated up to three cycles. In each cycle samples were frozen for at least 12 h at −80 ± 10°C. Freezer stability of Prasugrel in rat plasma was assessed by analyzing the QC samples stored at −80 ± 10°C for at least 15 days. Samples were considered to be stable if assay values were within the acceptable limits of accuracy (i.e., ± 15% S.D.) and precision (i.e., ± 15% R.S.D.)
Application to a pharmacokinetic study in rats
In vivo oral pharmacokinetic study was performed in male wistar rats (n = 5, weight range 200 – 220 g) to demonstrate the applicability of newly developed and validated bioanalytical method using UPLC. Prasugrel was administered orally at a dose of 30 mg/kg in 0.25% sodium Carboxy Methyl Cellulose (CMC) suspension. Blood samples were collected from the retro-orbital plexus of rats under light ether anesthesia into microfuge tubes containing heparin as an anti-coagulant at 0.5, 1, 2, 3, 4, 5, 8, 10, 12 and 24 h post-dosing. Plasma was harvested by centrifuging the blood at 2500×g for 5 min at 20°C and stored frozen at −80 ± 10°C until analysis. Rat plasma (100 μL) samples were spiked with IS and processed as described above and data was accepted based on the performance of QCs prepared using rat blank plasma (four QCs each at three concentration levels). The criteria for acceptance of the analytical runs encompassed the following: (i) not more than 33% of the QC samples were greater than ± 15% of the nominal concentration; (ii) not less than 50% at each QC concentration level must meet the acceptance criteria. Plasma concentration-time data of Prasugrel was analyzed by non-compartmental method using WinNonlin Version 5.1 (Pharsight Corporation, Mountain View, USA).
Application to In vitro metabolic stability
The metabolic stability of Prasugrel was performed using human liver Microsomes. The triplicate glass tubes contain reaction volume of 500 μL. Incubation of Prasugrel with human liver Microsomes was carried out at 37°C in a bench-top Lab-Line shaker. The incubation solution contained 100 mM potassium phosphate buffer (pH 7.4), 0.5 mg protein/mL microsomes, 5 mM MgCl2, 2 mM NADPH and 50 μM Prasugrel in a final volume of 500 μL. The enzyme reaction was initiated by adding NADPH after an initial 10-min pre-incubation. The reaction was terminated by adding 2 mL of ice cold ethyl acetate (contain 200 ng/mL I.S) at different interval 0, 15, 30, 45, 60, 90 & 120 minutes. The solution was vortex-mixed and centrifuged at 4°C for 10 min at 3500 rpm. The supernatant was transferred to a test tube and evaporated to dryness under vacuum in speedvac concentrator. The residue was reconstituted in 100 μL of mobile phase and 5 μl of this solution was injected to UPLC. Percent drug remaining versus time graph were plotted & resulting parameters like Rate Constant (K) & half life (t1/2) were determined using graph pad prism version 5.1.
Statistical analysis
Statistical analysis was performed by one-way ANOVA followed by Tukey’s test using Graph Pad prism software. All experimental values are expressed as mean ± Standard Deviation (SD). A value of P < 0.05 was considered significant. The pharmacokinetics parameters were determined non-compartmental method using WinNonlin Version 5.1 (Pharsight Corporation, Mountain View, USA).
Results & Discussion
Development and Optimization of the UPLC Method
Primarily, the drug was analyzed on a BEH C8 column using acetonitrile: water (50:50) as mobile phase at a flow rate of 0.15 mL/min. and a column temperature of 25°C. Under these conditions, the shape of the drug peak was not acceptable. Subsequent trials were made on plasma samples using different amounts of acetonitrile, pH and temperature. The peaks for this drug and the degradation products were not well separated or did not have an acceptable shape at column temperatures < 30°C and acidic pH. The best separation was achieved on the same column at 30°C using the mobile phase acetonitrile: water (25:75) in an isocratic mode. The flow rate was kept at 0.3 mL/ min at constant volume 3 μL and the detection wavelength was 240 nm.
Validation procedures of developed method
Recovery
The results of the comparison of pre-extracted standards versus post-extracted plasma standards were estimated for Prasugrel at 0.2, 2 and 15 μg/mL and the absolute percent mean recovery for Prasugrel at 0.2, 2 and 15 μg/mL were found to be 95.00%, 101.55% & 97.65% respectively. The absolute recovery of IS at 2 μg/mL was 90.35%.
Matrix effect, specificity and selectivity
In this study, the matrix effect was evaluated by analyzing QC low (0.2 μg/mL), QC medium (2.0 μg/mL) and QC high samples (15 μg/mL). Average matrix effect values obtained were 10.8, 12.8 and 8.8% at QC low, QC medium and QC high, respectively. Matrix effect on IS was found to be 4.6% at tested concentration of 2 μg/mL.
The specificity and selectivity has been studied by using independent plasma samples from six different rats. The method proved to be specific to each peak, which was indicated through purity data (Table 1, peak angle < peak threshold, it means peak is pure) determined through use of a PDA detector. Also, the resolution factor among various peaks was found to be >1.5, proving good separation among all the peaks. The method was also selective to each peak as all the peaks were pure, which was proved through PDA purity studies. The data of peak purity with resolution value are shown in Table1.
Table 1: Retention times and relative retention times of various peaks with their peak purity data.
| PEAKS | UPLC | ||||
|---|---|---|---|---|---|
| Retention Time (RT) | Relative Retention Time (RRT) | Peak purity angle | Peak threshold | Resolution factor | |
| Prasugrel | 0.61 | 1 | 2.9 | 4.1 | 3.1 |
| Clopidogrel | 0.41 | 0.67 | 2.2 | 3.3 | 3.7 |
| Morin | 1.5 | 2.45 | 2.9 | 3.5 | 3.5 |
RT = Retention time (minutes)
RRT = Relative retention time
Calibration curve and sensitivity
The linear regressions of the peak area ratios versus concentrations were fitted over the concentration range 0.1 – 20 μg/mL for Prasugrel in rat plasma. A typical equation of the calibration curve was: y = 0.0754x + 0.0021, r² = 0.9977, where y represents the ratios of Prasugrel peak area to that of IS and x represents the plasma concentration. The LLOQ for the determination of Prasugrel in plasma was 0.1 μg/mL. The precision and accuracy at LLOQ were 9.2 and 90.2% respectively.
Precision and Accuracy
The precision of the method was determined by calculating RSD for QCs at three concentration levels over three validation days. The Intra-day and inter-day precision was found to be range of 0.87 - 3.52 and 1.13 - 8.50 respectively at all QC level (0.2, 2, and 15 mg/mL). The accuracy of the method ranged from 90.33 to 101.54% at each QC level. Assay performance data are presented in Table 2. The above results demonstrate that the values are within the acceptable range and the method is accurate and precise.
Table 2: Precision and accuracy for Prasugrel of quality control sample in rat plasma (n = 5).
| Nominal Concentration (μg/mL) | Observed Concentration Mean ± SD | Precision (%)a | Accuracy (%)b | |
|---|---|---|---|---|
| Intra-day | ||||
| 0.2 | 0.190 | ± 0.002 | 0.991 | 95.167 |
| 2 | 2.031 | ± 0.072 | 3.526 | 101.533 |
| 15 | 14.648 | ± 0.128 | 0.872 | 97.656 |
| 0.2 | 0.181 | ± 0.015 | 8.507 | 90.333 |
| 2 | 2.031 | ± 0.144 | 7.090 | 101.543 |
| 15 | 14.648 | ± 0.167 | 1.138 | 97.656 |
aExpressed as % R.S.D. = (S.D/mean) × 100.
bCalculated as (mean determined concentration/nominal concentration) × 100.
Table 3: Summary of stability of Prasugrel under various storage conditions (n = 5).
| Condition | Spiked concentration | found concentration | RSD (%) | RE (%) |
|---|---|---|---|---|
| Ambient, 2 h | 0.2 15 |
0.184 14.548 |
6.530 0.604 |
-7.833 -3.013 |
| -20°C, 30 days | 0.2 15 |
0.190 14.598 |
10.500 1.360 |
-5.000 -2.678 |
| Three freeze–thaw | 0.2 15 |
0.198 14.981 |
12.392 3.596 |
-1.000 -0.124 |
| Autosampler ambient 24 h | 0.2 15 |
0.182 14.665 |
8.594 1.033 |
-9.000 -2.233 |
| 10 days -80°C | 0.2 15 |
0.186 14.835 |
9.614 3.931 |
-6.833 -1.102 |
RE % = (observed-nominal)/nominal X 100
Stability
The stability results showed that Prasugrel spiked into rat plasma was stable for 2 h at room temperature, for 30 days at -20°C, and during three freeze–thaw cycles. Stability of Prasugrel extracts in the sample solvent on an auto sampler was also observed over a 24 h period. The long-term stability was assessed after storage of the standard spiked plasma samples at -20°C for 30 days. The results of stability experiments are listed in Table 3.
Pharmacokinetics studies
The pharmacokinetic results are shown in Figure 2 and its parameters are summarized in Table 4. The statistical comparison of mean plasma concentration of Prasugrel in three groups by one-way ANOVA followed by Tukey’s test revealed significantly higher (P < 0.01) plasma. The level of prasugrel from 2 to 24 h in morin-pretreated group in comparison with the control and morin co-administered groups. However, no significant change (P > 0.05) was observed in plasma concentration of Prasugrel in morin co-administered group at all the studied time points in comparison with that of control group. Further, analysis of pharmacokinetic parameters revealed that morin pretreatment caused a significant elevation in Cmax (1.4 fold) of Prasugrel in comparison with the control group and co-administered groups at 0.5 hr as shown in Figure 2. As the results shown in Table 4, the elimination half-life (t1/2), AUC0-t, Clearance (CL), Mean Residential Time (MRT) also affected with respect to time in comparison at different groups (control, co-administered & pre-treated).
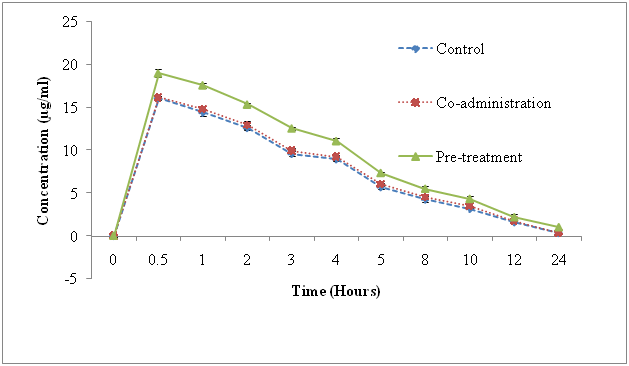
Figure 2: Mean plasma concentration–time profiles after the oral administration of prasugrel (control, 30 mg/kg), co-administration (prasugrel + morin) & pre-treatment Bars represent the standard deviation (n = 5).
Table 4: Pharmacokinetic parameters after oral administration of single dosage 30 mg/kg Prasugrel to rats (n = 6).
| Parameters | Control Mean ± SD | Morin (co-administered) + PCM | Morin (pretreatment) + PCM |
|---|---|---|---|
| Mean of observed concentration N = 3 | |||
| Cmax (μg/ml) | 16.112 | 16.218 | 18.994 |
| Tmax (h) | 0.500 | 05.000 | 0.500 |
| AUC0-t(μg h/Ml) | 95.104 | 97.115 | 125.221 |
| AUC0-∞(μg h/Ml) | 97.001 | 98.211 | 130.567 |
| CL (L/h/kg) | 00.621 | 00.658 | 00.455 |
| t1/2 (h) | 07.574 | 07.998 | 09.862 |
| MRTt | 06.112 | 06.554 | 07.236 |
| % RB | 100 | 101.24 | 134.60 |
Relative bioavailability (RB) % = AUC co-admin or pre-treated/ AUC control X 100.
In vitro metabolic stability
Metabolic stability has been defined as the percentage of parent compound lost over time in the presence of the test system. Because a majority of drug metabolism occurs in the liver, the in vitro human liver microsomes have been used to evaluate metabolic stability. By understanding the metabolic stability of compounds early in discovery, the potential for a drug candidate to fail in development as a result of pharmacokinetic reasons may be reduced. Microsomes may be more applicable because of the low volumes used in these assays and to achieve goal is to screen Prasugrel in a high-throughput manner. The metabolic stability of Prasugrel is determined using human liver Microsomes in an in vitro design. Approximately, 65% of the intact Prasugrel remained in the incubation mixture over the time of 120 minutes as shown in Figure 3. This indicates that the drug Prasugrel was significant metabolize and instable in human liver Microsomes up to 2 hrs at 37 ± 1°C with half life of 78.31 hr. The non-linear regression analysis of the data showed good fitting with mono exponential decay followed by degree of freedom of 18 were calculated by graph pad prism software. The determined parameters are rate constant (K min-1), half life (t1/2 mins), Degree of Freedom (df) & regression co-efficient (r²) shown in Table 5.
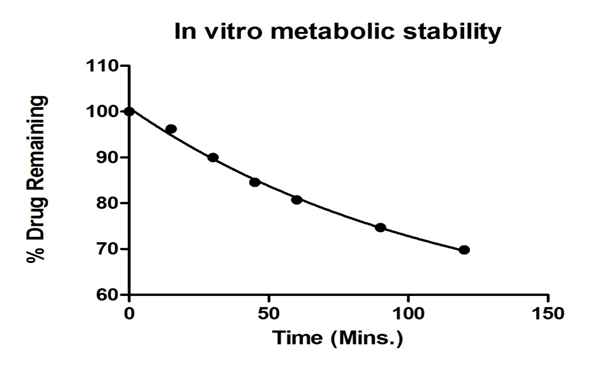
Figure 3: Plot of parent drug remaining in the incubation mixture versus time of prasugrel.
Table 5: Metabolic stability data for Prasugrel.
| parameters | Observed values |
|---|---|
| K (min-1) | 0.0088 |
| t1/2 (minutes) | 78.31 |
| R² | 0.995 |
| Df (degree of freedom) | 18 |
Conclusion
The method proved to be simple, accurate, faster, precise, specific and selective. Further, the present study suggests that there might be a potential interaction between Prasugrel and Morin and therefore, quantitative evaluation of Prasugrel–morin interaction in humans needs to be verified to avoid food–drug interactions. The pre-treatment of naturally occurring dietary supplement morin significantly increased the relative bioavailability of prasugrel. It is hoped that this report on development of a method for analysis of Prasugrel will be helpful for drug-flavonoid interactions and Cytochrome induction & inhibition, phenotyping studies around the world by saving them from unnecessarily performing similar studies.
Acknowledge:
The authors are thankful to Vice Chancellor, Jamia Hamdard, New Delhi for his constant encouragement and support. We also acknowledge Department of Science & Technology (DST) for providing research fellowship to Dr. Kapendra Sahu.
Conflict of interest:
The authors declare that they have no conflict of interest.
References
- 1. Rigobello, C., Barden, A.T., Steppe, M. LC method for determination of prasugrel and mass spectrometry detection for thermal and alkaline degradation products. (2015) Pharmazie 70(8): 515-521.
Pubmed || Crossref || Others - 2. Singh, D.K., Sahu, A., Handa, T., et al. Study of the forced degradation behavior of prasugrel hydrochloride by liquid chromatography with mass spectrometry and liquid chromatography with NMR detection and prediction of the toxicity of the characterized degradation products. (2015) J Sep Sci 38(17): 2995-3005.
Pubmed || Crossref || Others - 3. http://en.wikipedia.org/wiki/Prasugrel. (accessed on 12.02.17).
Crossref || Others - 4. Yu, K.S., Park, K.W., Kelly, R.P., et al. Pharmacokinetic and pharmacodynamic effects of prasugrel in healthy Korean males. (2013) J Cardiovasc Pharmacol 62(1): 72-77.
Pubmed || Crossref || Others - 5. Farid, N.A., McIntosh, M., Garofolo, F., et al. Determination of the active and inactive metabolites of prasugrel in human plasma by liquid chromatography/tandem mass spectrometry. (2007) Rapid Commun Mass Spectrom 21(2): 169-179.
Pubmed || Crossref || Others - 6. Choi, B.C., Choi, J.S., Han, H.K. Altered pharmacokinetics of paclitaxel by the concomitant use of morin in rats. (2006) Int J Pharm 323(1-2): 81-85.
Pubmed || Crossref || Others - 7. Sahu, K., Shaharyar, M., Siddiqui, A.A. Effect of Morin on pharmacokinetics of Piracetam in rats, in vitro enzyme kinetics and metabolic stability assay using rapid UPLC method. (2013) Drug Test Anal 5(7): 581-588.
Pubmed || Crossref || Others - 8. Singh, S.P., Wahajuddin, M., Raju, K.S., et al. Reduced bioavailability of tamoxifen and its metabolite 4-hydroxytamoxifen after oral administration with biochanin A (an isoflavone) in rats. Phytother Res 26(2): 303-307.
Pubmed || Crossref || Others - 9. Wu, J. Cytochrome P450 Enzymes Biochemistry, Pharmacology and Health Implications. (2014) Nova Science Publishers.
Pubmed || Crossref || Others - 10. Rama Raju, K.S., Taneja, I., Singh, S.P., et al. Simultaneous determination of centchroman and tamoxifen along with their metabolites in rat plasma using LC-MS/MS. (2015) Bioanalysis 7(8): 967-979.
Pubmed || Crossref || Others - 11. Taneja, I., Raju, K.S., Challagundla, M., et al. LC-ESI-MS/MS method for bioanalytical determination of osteogenic phytoalexin, medicarpin, and its application to preliminary pharmacokinetic studies in rats. (2015) J Chromatogr B Analyt Technol Biomed Life Sci 1001: 9-16.
Pubmed || Crossref || Others










