
Ethacrynic Acid and Ciclopiroxolamine in Various Cancer Cells
Nkem Purity C. Emekwue1,2, Hans Weiher2Ingo G.H. Schmidt-Wolf1*
Affiliation
1Center for Integrated Oncology (CIO), University Hospital Bonn, Bonn, Germany
2Bonn-Rhein-SiegUniversity of Applied Sciences, Rheinbach, Germany
Corresponding Author
Prof. Dr. Ingo G.H. Schmidt-Wolf, Center for Integrated Oncology (CIO), University Hospital Bonn, Sigmund-Freud-Straße 25, 53105 Bonn, Germany, Tel: +49 228 287 17050/ Fax: +49 228 287 9080059; E-mail: ingo.schmidt-wolf@ukbonn.de
Citation
Schmidt-Wolf, I.G.H., et al. Ethacrynic Acid and Ciclopiroxolaminein Various Cancer Cells. (2018) Int J Hematol Ther 4(1): 16-24.
Copy rights
© 2018 Ingo Schmidt-Wolf, G.H. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Wnt pathway; Beta-catenin; Ethacrynic acid; Cicloproxolamine
Abstract
Extensive data from both in vitro and animal studies have demonstrated the potency of a deregulated activated Wnt pathway. Herein, we report the antitumor effects of already established novel Wnt pathway inhibitors – ethacrynic acid (EA) and Ciclopiroxolamine (CIC) on various cancer cell lines using the WST-8 assay. Our results demonstrate the significant cytotoxic potency of EA and CIC in vitro and display their strong efficacy on various tumor cells. Of particular interest, colon cancer was the most significantly affected by both drugs of all the tumor types tested in this study. This study provides a strategy for targeting Wnt-driven cancers and highlights those cancers that show a higher susceptibility to the cytotoxic effects of the Wnt inhibitors as potential candidates for further study.
Introduction
The challenge in cancer management worldwide, despite significant progress in therapy and remission rates, has propelled research to establish the therapeutic relevance of cancer stem cells (CSCs) as they play a key role in drug resistance, progression and recurrence of several cancers. Hence, research interests and efforts are currently geared towards targeting CSCs as a panacea to the menace. The Wnt signaling pathway apart from its role in tissue regeneration has been revealed as a major driver of cancer stem cell proliferation[1,2]. Most current anti-cancer drugs, small molecule inhibitors and monoclonal antibodies (mAbs), are designed to target rapidly proliferating cells which represent committed cancer cells but not CSC[3]. Today, since current cancer therapies fail to eradicate CSCs, the goal of current cancer therapeutics research is focused on targeted therapies in combination with standard chemotherapies. Therefore, a selective targeting of the Wnt signaling pathway has been considered a promising strategic advantage for therapy as several preclinical experiments have demonstrated that inhibition of this pathway would not only diminish cancer stem cell pool but also eradicate cancer.
The Wnt pathway is an extensively studied and highly evolutionarily conserved pathway that plays a crucial role in animal embryonic development, during which it has numerous roles including cell proliferation, survival, migration and polarity, specification of cell fate, and self-renewal property[4]. In the healthy adult tissues however, it is largely inactive due to the degradation of β-catenin, the hallmark effector of the canonical Wnt pathway. The pathway is driven by Wnt ligands (WNT), which are a family of 19 secreted cysteine-rich glycoproteins that activate receptor-mediated signaling pathways – both canonical (β-catenin dependent) and non-canonical (β-catenin independent) mechanisms. Normally in the absence of stimulation, the cytoplasmic pool of β-catenin is tightly regulated and maintained at a low level through ubiquitin-proteasome-mediated degradation via phosphorylation by the multiprotein “destruction complex”. The destruction complex is composed of glycogen synthase kinase 3 β (GSK-3 β), casein kinase 1α, the scaffold protein Axin, and the tumor suppressor adenomatous polyposis coli (APC), among others[5].
Upon Wnt signaling, the secreted Wnt proteins bind to a receptor complex, consisting of a member of the Frizzled (Fzd) family, and the low-density lipoprotein-receptor related proteins (LRP) 5 or LRP6. Subsequently, the cytoplasmic adaptor protein dishevelled (Dvl) is phosphorylated and inhibits glycogen synthase kinase (GSK)-3β activity through its association with Axin. Unphosphorylated β-catenin accumulates in the cytoplasm and is translocated into the nucleus, where it exerts its downstream effects by mediating LEF/TCF dependent transcription of Wnt-target gene. To generate a transcriptionally active complex, β-catenin acts as a transcription activator by displacing Grouchos and recruiting the co-activators cAMP response element-binding protein (CBP) or its homolog p300 along with other components of the basal transcription machinery (such as CtBP, Foxo, TNIK, Bcl9, and Pygopus)[6] thus activating the transcription of Wnt target genes such as c-myc and cyclin D1, that are involved in developmental stages and adult tissue homeostasis[7]. Anomalous activation of the Wnt pathway is often associated with a host of oncogenic effects[8-10].
Consequently, with the growing popularity of drug repositioning to improve therapeutic effectiveness, this project joins a stream of several other researches geared towards therapeutic modulation of signaling pathways using repurposed drugs. In a series of studies, our group already successfully demonstrated this strategy by targeting the Wnt pathway in different cancer types. Recently we confirmed that the diuretic agent ethacrynic acid (EA) and the antifungal agent (CIC) ciclopiroxolamine (CIC) inhibit Wnt/β-catenin signaling[11]. In addition, we demonstrated through several experiments that these re-purposed drugs, naftifine, flunarizine, piroctoneolamine, among others target the Wnt/β-catenin pathway and exhibit cytotoxic effects against a wide range of hematological neoplasms[11-13].
Our study therefore aimed to examine the cytotoxic effect of the Wnt inhibitors, EA and CIC on the viability of various cancer cell lines and determine which of the cancer types is most susceptible to the killing effect of either or both drugs.
Materials and Methods
Cell lines and culture conditions
All cell lines were obtained from DSMZ (Braunschweig, Germany) or ATCC (LGC Standards, Wesel, Germany), prepared in their respective standard media and incubated aseptically at 37°C in the presence of 5% CO2 and 80% humidity. The pancreatic cancer cell line DanG; prostate cancer cell line DU145 and ovarian cancer cell line A278003 were cultured in 50 % Roswell Park Memorial Institute (RPMI) medium (PAN Biotech, Aidenbach, Germany) supplemented with 50% heat inactivated fetal calf serum (FCS) (Gibco Life Technologies, Darmstadt, Germany) and 1% penicillin / streptomycin (P/S) (Life Technologies, Darmstadt, Germany). The colon cancer cell lines; Col 205 and Col 201 were cultivated in 90% RPMI medium plus 10% FCS and 1% P/S. The kidney cancer cell lines; A498 and A704 and the colon fibroblast cell line CCD18Co were cultured in 50% Eagle’s Minimum Essential Medium (EMEM) supplemented with 50% FCS plus 1% P/S. The melanoma cancer cell line HT-144; breast cancer cell line SKBR-3 and bladder cancer cell line T24 were cultured in 50% McCoy (modified) medium supplemented with 50% FCS plus 1% P/S. The bronchial cancer cell line A549 was grown in 50% Ham’s F-12K (Kaighn’s) medium supplemented with 50% FCS plus 1% P/S. The breast cancer cell lines MCF-7 and T47D were maintained in 50% EMEM and RPMI media respectively supplemented with 50% FCS, 1% P/S and 10 μg ml-1 insulin (Invitrogen). Confluent cultures were either split 1:4, 1:3 or 1:2 every 2 - 3 days. Cells were harvested by using 0.25% or 0.05% trypsin – EDTA solution (Gibco – Life Technologies, Darmstadt, Germany), centrifuged at 1200 rpm for 8 minutes and resuspended in 1ml media to define the cell count. Medium was renewed every 2 to 3 days.
Drug solutions
The drugs ethacrynic acid (EA) and ciclopiroxolamine (CIC) both procured from (Sigma - Aldrich, Steinheim, Germany) were dissolved in 100% ethanol. Aliquots of EA were prepared in PBS at a concentration of 30 μM, 50 μM, 100 μM and 200 μM. CIC was prepared at working concentrations of 5μM, 10 μM, 50 μM and 100 μM. The sub confluent cell lines (70 - 80% confluent) were treated with varying drug concentrations with an incubation time of 48 h. The inhibitory concentration 50 (IC50) of the drugs found from previous research formed the basis for the choice and wide concentration range utilized for this study. This was necessary considering real – life therapeutic interventions which would require low dosage to produce the required effects in patients.
Cell viability and cytotoxicity assay
Cell viability was assessed by WST - 8 staining with the Cell Counting kit - 8 (Dojindo, Shanghai, China). This assay is based on the ability of dehydrogenase enzymes in metabolically active cells to reduce the high water - soluble tetrazolium salt WST - 8 to an orange - colour formazan dye, which is soluble in the tissue culture medium. The amount of the formazan dye generated is directly proportional to the number of living cells. After an incubation time of 1 hour, the absorbance was measured at 450 nm.Cell lines were seeded in 96 - well plates at a density of 1 x 104 cells / well in 100 μl medium in triplicates under standard culture conditions and incubated for 24 h. This was followed by treatment with the drug agents (EA and CIC) in a range of concentrations and incubated for 48 h. A quadruplet of negative control was made for each cell line without any drug agent. The same was provided for medium controls without any cells and drugs which were measured and used for final evaluation.
Data analysis
Data from the WST assay was analyzed using Microsoft Excel, graphs were plotted with Qtiplot and exported to Inkscape for graphic illustrations. The cell viability for each well was calculated using the following formula:
Absorbance of treated samples – Average absorbance of blank x 100 %
Average absorbance of negative control – Average absorbance of blank
Data are shown as mean ± standard error of the mean (SEM). Statistical analysis was done using the R programming language (ref). One - way ANOVA was used to test for significance between treatment groups. Post-hoc pairwise comparisons of means were done using t tests on pooled SD. The p values were adjusted by the holms method. A p–value of less than 0.05 was considered significant.
Results
Effect of CIC and EA on the viability of Colon fibroblast cell line CCD-18Co
We first wanted to verify whether the utilized drug concentrations were not significantly toxic to our control cell line colon fibroblast cells CCD-18Co so as to exclude drug-induced cytotoxicity as the primary reason for decline in cell viability during subsequent analysis of our test cancer cell lines. The results obtained indicate a non-significant decline in CCD-18Co cell viability irrespective of the concentrations of both drugs (Figure 1). Interestingly, we notice that CIC showed a statistically significant toxicity only at the highest tested dose of 100 μM (p-value < 0.05). However, according to the range of concentrations used in this study, it reveals the potential harmful effect of CIC even on healthy cells at lower concentrations (cell viability dropped to about 90% at 10 μM) in comparison to higher doses of EA.
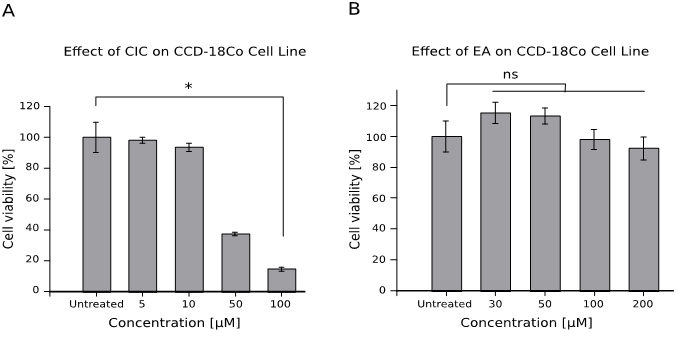
Figure 1: Control Cell line – Colon fibroblast CCD-18Co survive drug treatment
1 x 104 cells were treated with 10 μl of various concentrations of CIC and EA for 48 hours. Cell viability was performed afterwards using the WST assay. Mean cell viability is given as mean ± standard error of the mean (SEM). Data represents an experiment performed in quadruplets out of two independent experiments with similar results.
Effect of CIC and EA on the viability of prostate cancer cell line DU145
The result (Figure 2) shows that both drugs reduced the viability of prostate cancer cell lines, although CIC killed nearly all the cells at all doses (p < 0.001).
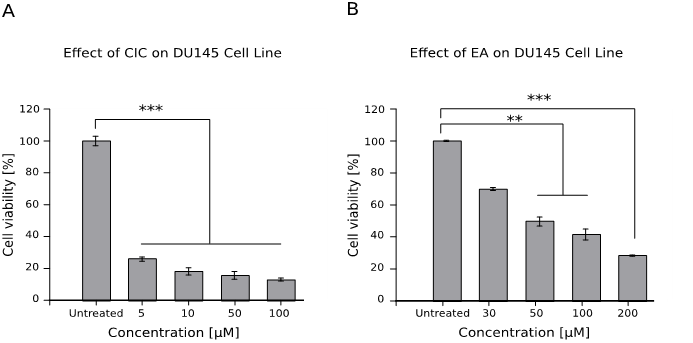
Figure 2: Effect of CIC and EA on the viability of prostate cancer cell line DU145
1 x 104 cells were cultured for 48 hrs with different concentrations of both drugs and viability was assessed afterwards using the WST assay. Results are shown as mean ± standard error of the mean (SEM) and represent data from three independent experiments performed in quadruplets.
Effect of CIC and EA on the viability ofcolon cancer cells Col 201 and Col 205
Both CIC and EA induced a significantly higher decrease in viability of colon cancer cell lines. We observed that the EA treated Col 201 cells (Figure 3B) share the same pattern of toxicity with Col 205 cells treated with CIC (Figure 3C) as the drugs nearly killed all cells (p < 0.001). In Figure 3D, we could observe a dose dependent decrease in viability; the lowest dose of 30 μM yielded no significant decrease in viability while there was significant effect for higher doses.
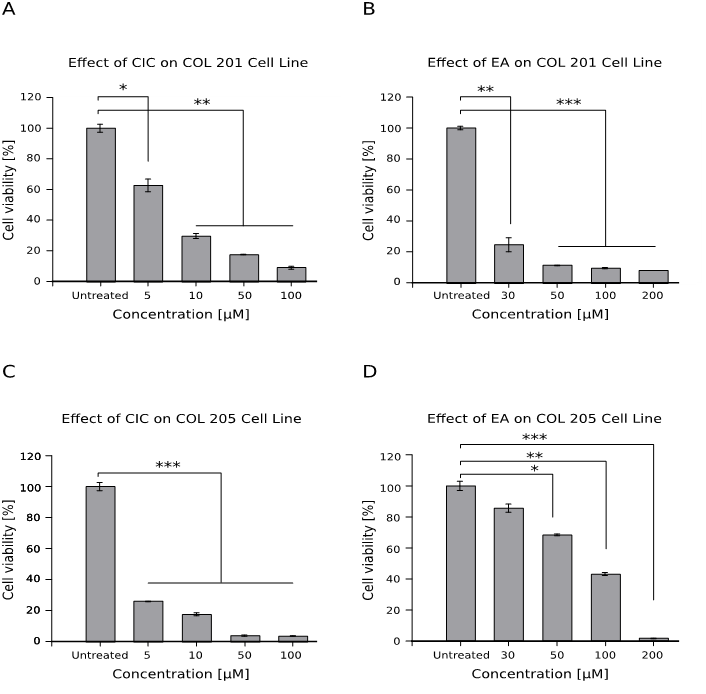
Figure 3: Effect of CIC and EA on the viability of colon cancer cells – Col 201 and Col 205
1 x 104 cells of two different colon cancer cell lines were cultured for 48hrs with different concentrations of both drugs and viability was assessed afterwards using the WST assay. Cell viability is given as mean ± standard error of the mean (SEM) from quadruplet wells out of two independent experiments.
Effect of CIC and EA on the viability of ovarian cancer cell line A278003
Both drugs showed a strong cytotoxic effect on the viability of ovarian cancer cells at all tested doses (p < 0.001, see Figure 4).
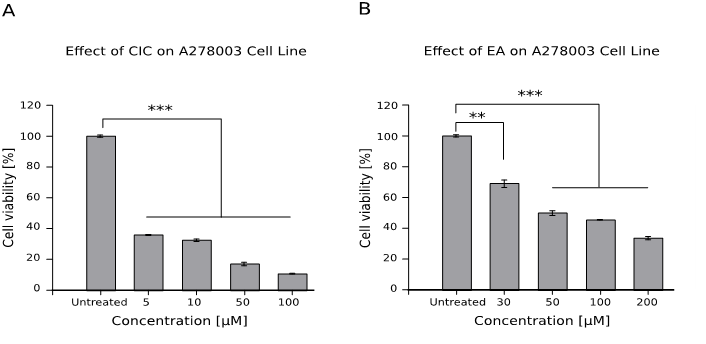
Figure 4: Effect of CIC and EA on the viability of ovarian cancer cell line A278003
1 x 104 cells were cultured for 48hrs with different concentrations of both drugs and viability was assessed afterwards with the WST assay. Results are shown as mean ± standard error of the mean (SEM) and represent data from two independent experiments performed in quadruplets.
Effect of CIC and EA on the viability of melanoma cell line HT-144
Figure 5B shows a statistically significant decrease in cell viability from the concentrations 50 μM to 200 μM (p < 0.001). However, compared to the untreated cell, CIC (Figure 5A) statistically decreased viability from the lowest concentration of 5 μM (p < 0.01) and even more so for 10 μM to 100 μM (p < 0.001).
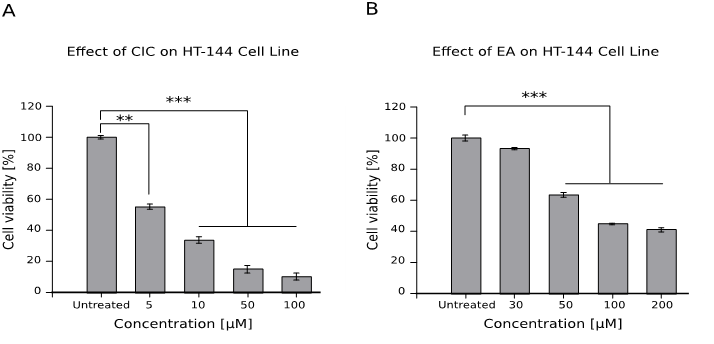
Figure 5: Effect of CIC and EA on the viability of melanoma cell line HT-144
1 x 104 cells were cultured for 48 hrs with different concentrations of both drugs and viability was assessed afterwards with the WST assay. Results are shown as mean ± standard error of the mean (SEM) and represent data from three independent experiments performed in quadruplets.
Effect of CIC and EA on the viability of pancreatic cancer cell line DanG
Both drugs reduced cell viability (Figure 6), however, the lowest tested doses of EA 30 μM and 50 μM produced minimal toxicity compared to the lowest tested doses of CIC 5 μM and 10 μM.
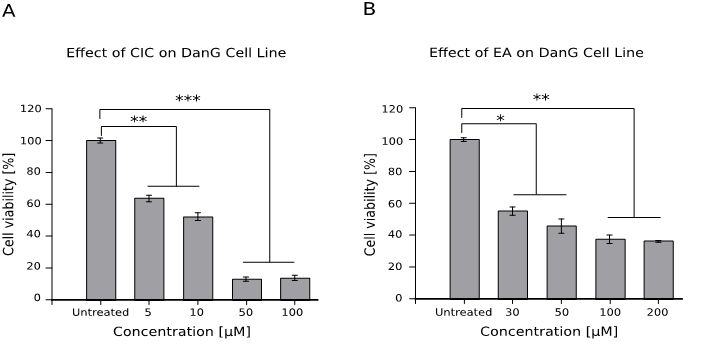
Figure 6: Effect of CIC and EA on the viability of pancreatic cancer cell line DanG
1 x 104 cells were cultured for 48hrs with different concentrations of both drugs and viability was assessed afterwards with the WST assay. Results are shown as mean ± standard error of the mean (SEM) and represent data from three independent experiments performed in quadruplets.
Summary of cell viabilities of cancer cells at 10 μM CIC and 200 μM EA
An overview of the viability of all cancer lines tested in this study was made at 10 μM of CIC and at 100 μM of EA to compare the degree of susceptibility of the cancer cells to the cytotoxic effect of both drugs at the selected doses (Figure 7). Here, almost all cell lines show a viability of less than 60% in response to both drugs. However, compared to EA, CIC appears to have the most significant toxic effect on all cells with 10 of the 13 cancer cell lines having viability below 40% (Figure 7 A). The control cell line (colon fibroblast) however, survived treatment. The cancer cell lines; prostate, bladder and colon cancer col 205 are the most affected by CIC, with all three having almost the same percentage viability at nearly 20% while EA killed mostly both renal, col 201 and breast SKBR-3 cells. Compared to others, it is seen that bladder and bronchial cancers showed lesser response to the cytotoxic effect of EA by having almost the same viability of 54.4 % and 53.4 % respectively (Figure 7 B). Taken together, we have tested two anti-tumor drugs (CIC and EA) on different cancer cell lines and demonstrated that colon cancer was the most significantly affected by both drugs of all the tumor types tested in this study while bronchial, breast MCF-7 and pancreatic cancer cells were the least affected by both drugs.
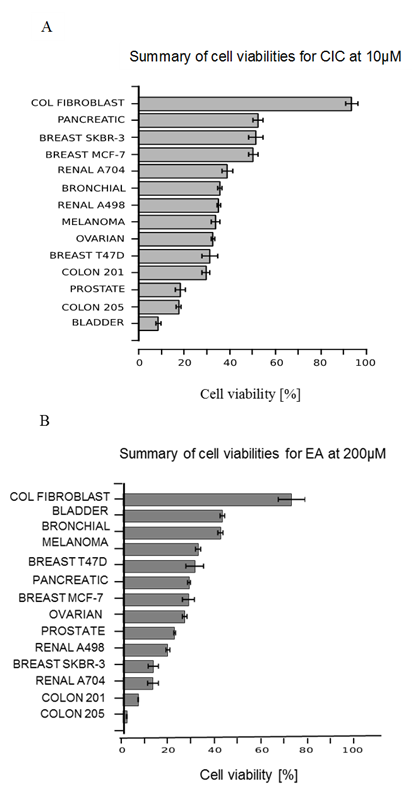
Figure 7: Summary cell viabilities of all cancer cells at 10 μM CIC and 200 μM EA
A summary graph comparing the degree of susceptibility of the different cancer cell lines to the killing effect of CIC at 10 μM and EA at 200 μM after 48 hrs.
Discussion
Targeted therapies involving signaling pathways in cancer are emerging as promising alternatives; but malfunction of the Wnt/β-catenin has been implicated in many oncogenic transformations[14]. Indeed multiple attempts to develop inhibitors against key Wnt co-receptors, including LRP6 and FZD, have been reported[15,16] with varying degrees of success. Recently, there has been much interest in the exploration of relatively repurposed drugs with anti - tumor potential and repositioning of existing drugs to win the battle against cancers, especially Wnt-dependent cancers. Especially in oncology, where there is an ever-increasing demand for new therapies, drug repositioning could offer a faster and economically more interesting way of fighting this class of disease. In several studies, the repurposed drugs - ethacrynic acid (EA) and ciclopiroxolamine (CIC), revealed anti carcinogenic effects in vitro and have been identified as Wnt inhibitors[10] in lymphoma and melanoma cells. The present study in showing reduction of proliferation of cancer cell lines with the use of cell proliferation assay suggests that these novel Wnt inhibitors may be used to achieve significant reduction in cell viability across a wide array of tumor types. EA and CIC have been shown to share a common property of influencing the expression or stabilization of β-catenin – the key protein of the canonical Wnt pathway, even though both drugs do this through different mechanisms.
Previous investigations of our group revealed both EA and CIC to be potent inducers of apoptosis in cancer cells. EA, as stated in several prior studies, does not only act as a loop diuretic agent by inhibiting the Na+-K+-2Cl- kidney import but has also been found to possess cytotoxic ability towards different cancer cells. The biological effect of EA is in the modulation of detoxification gene expression (inhibition of glutathione-S-transferase), causing increased cellular oxidative stress due to elevated glutathione (GSH) levels thus inducing apoptosis. Possible induction mechanisms include the alteration of sulfhydryl status by the electrophilic properties of EA or by elevations of endogenously generated oxidative stress via transient removal of GST pi from the cytosolic GST pool[17]. However, Aizawa et al., in their study did not confirm this finding, demonstrating there is an independence between GSH levels and EA related induction of apoptosis[18]. A recent study showed that EA and its butyl ester prodrug induced apoptosis in leukemia cells through a hydrogen peroxide–mediated pathway[19]. Because EA can bind to any free thiol, it is likely that the mechanism of EA-induced cell death is variable and will depend upon cell type and context. Furthermore, as an antagonist of the Wnt/β-catenin signaling pathway, EA suppresses cell survival by selectively inhibiting the recruitment of lymphoid enhancer-binding factor-1 (LEF1), a key transcription factor of this pathway which helps to regulate genes involved in tumor cell death mechanisms[10]. Dysregulation of LEF1 has been identified in several cancers including chronic lymphocytic leukemia (CLL) and high expression is associated with poor survival of patients[20]. It has been demonstrated that by interacting with LEF-1, EA subsequently de-stabilizes the LEF-1/β-catenin complex. It also alters the expression of the Wnt target genes cyclin-D1, fibronectin and crucial proteins of the Wnt pathway. The cytotoxic mechanism of EA was revealed in a study by Kim et al., 2012 where EA inhibited the Top Flash reporter (TCF/LEF) construct in a dose - dependent manner and was further demonstrated by co-immunoprecipitation studies to target LEF-1, destabilize formation of its complex with β-catenin and consequently inhibiting the Wnt pathway[21]. In a previous study we showed that EA caused a dose-dependent decline in the expression of three Wnt target genes, LEF-1, cyclin D1 and fibronectin, reflecting EA inhibition of Wnt/β-catenin signaling in chronic lymphocytic leukemia cells[5]. Another study has additionally shown that treatment of myeloma cells with EA results in decreased levels of β-catenin, which points toward existence of several inputs of this drug in Wnt signaling inhibition. In vivo, EA alone has shown excellent promise and was able to inhibit myeloma growth and prolong survival in mice more efficiently than lenalidomide, the current drug of choice for patients with multiple myeloma[11]. In humans the maximum dose of EA when administered by intravenous injection is 100 mg/day, which results in plasma levels of around 30 μM[22]. This corresponds to the Wnt inhibitory doses used in the in vitro myeloma studies[21,23]. In mice, the oral dose of 450 μg/day should result in plasma levels close to those in humans mentioned above; meaning that inhibition of tumor growth may also be feasible in humans. However, no reports of such a study currently exist. EA is a weak anti-tumor agent and has been demonstrated to exert its anti-proliferative effects against human cancer cells but only at higher concentrations (60 – 100 μM)[24]. The relative lack of potency and its diuretic property has limited EA from serving as an effective chemotherapeutic agent[25].
In addition, other signaling pathways may also contribute to the cytotoxic effects of EA on various cell types. In their report, Han et al., showed that EA could inhibit activation of the NF-κB pathway at multiple steps[19]. Previous studies have revealed that NF-κB signaling is another anti-apoptotic pathway which is constitutively activated in CLL cells and inhibition of NF-κB by drugs induces apoptosis of CLL cells[26,27]. Thus, inhibition of NF-κB may synergize with Wnt antagonism to impair CLL survival. It has also been reported that the mitogen activated protein kinase (MAPK) pathway may be involved in EA-induced cell death[18].
The Wnt pathway is not exclusively employed during development or over activated in cancer but also in adults where many healthy tissues rely on it for renewal and homeostasis maintenance, most notably the intestine, haematopoietic system, hair, bones and skin. Therefore, this poses a potential risk of adverse reactions in all these organ systems, which has indeed been observed for many Wnt - targeting compounds upon attempts to push them into the clinics. According to their study, Mook RA et al., reported failure of many anti Wnt agents for example; XAV939 and LGK974 cause severe intestinal toxicity in mice, while OMP18RP induces abdominal pain, constipation and diarrhea in patients[28]. This may explain, in part, the reason for the high level vulnerability of our tested colon cancer cell lines to the drugs. Col 205 cells (Fig 3B) were less sensitive to EA compared to Col 201 cells. At the lowest dose of 30 μM, EA reduced viability by 25% in Col 201 cells but Col 205 cells retained a viability of 87%. This raises the question whether Col 205 cells have a different surface structure which inhibits interaction with EA. Reports have shown that N-acetyl L-cysteine (NAC) could protect cancer cells from apoptosis upon exposure to EA by reacting with the α, β-unsaturated ketone in EA[18]. One possible explanation for the high sensitivity of Col 201 cells to EA may be either attributed to the multiple effects of EA on not only Wnt but other related pathways such as the NF-κB signaling or that Col 205 cells may have higher levels of NAC than Col 201. Further research is nevertheless needed to determine the level of NAC and identify the receptor or surface structure by which EA binds to cancer cells, in this case, colon cancer.
CIC is a synthetic antifungal agent used to treat cutaneous fungal infections for over two decades. Apart from its antimycotic activity, CIC also has a broad spectrum action against bacteria and yeast[29]. Its mechanism of action seem diverse; involving disruption of membrane function in fungi or targeting different metabolic and energy producing processes in bacteria[30]. CIC exerts its effect in the yeast Saccharomyces cerevisiae by blocking cell cycle progression[31], disrupting DNA repair and replication including some elements of intracellular transport, cell division signals and structures[32]. However, ciclopiroxolamine has also been shown to exhibit promising anti-cancer activity in preclinical models of several cancers[5] as has been found in its ability to inhibit the growth and viability of myeloma cells, acute myeloid leukemia (AML) cell lines and primary patient samples in vitro and in vivo, including the leukemic stem cell fraction[33]. In vitro analysis reveal that CIC induced cell cycle arrest at G1/G0 phase by upregulation of cyclin-dependent kinase (CDK) inhibitor p21 and down regulation of cellular expressions of cyclin D, retinoblastoma protein (Rb) and phosphorylated Rb (pRb)[34].
Mechanistically, both antimicrobial and anticancer activities of CIC are attributed to iron chelation[33]. There is often elevated levels of intracellular iron in cancer cells, and iron chelation appears to selectively target cancer cells over normal cells[35]. By binding intracellular iron, CIC, which inhibits iron – dependent enzymes such as ribonucleotide reductase and deoxyhypusine hydroxylase to disrupt cell cycle progression[33], disrupts iron-dependent cellular processes such as the mammalian target of rapamycin (mTOR), the eukaryotic translation initiation factor 5A (eIF5A) function[36] and Wnt signaling in tumor cell lines as well as leukemic blasts isolated from patients[37]. CIC also enhances apoptotic death by increasing caspase-3/7 activity and suppressing the expression of the anti-apoptotic proteins survivin and Bcl-xL in human rhabdomyosarcoma cells[29]. Apart from chemotherapy, there has been increasing evidence demonstrating the repositioning of CIC for treatment of other human diseases. CIC has been reported to ameliorate diabetes and its complications[38], inhibit the expression of HIV-1 gene to prevent HIV infection[39], serve as an alternative to recombinant vascular endothelial growth factor (VEGF) gene therapy for therapeutic angiogenesis[40] and improve age-associated cardiovascular defects[41]. CIC treated stem cells have been shown to have down regulated levels of proteins, which are involved in nucleotide biosynthetic process, cell proliferation, differentiation and development, signal transduction, G protein-coupled receptor endocytosis and gene expression[42]. In their work, Zhou and his colleagues found CIC induced autophagy in rhabdomyosarcoma (Rh30 and RD) cells, which was mediated by ROS induction, leading to activation of JNK cascade[43]. It is clear, therefore, that the antitumor mechanism of CIC is complex and needs to be further investigated.Through several experiments, our group has identified CIC as a potent inhibitor of the Wnt/β-catenin pathway which acts by targeting and attenuating cytoplasmic β-catenin levels. This approach has seen the depletion of survival of various Wnt-driven cancers including multiple myeloma, pancreatic and renal carcinoma[5,44,45]. In addition, we could also show that CIC offers a synergistic therapeutic effect upon co-administration with other anticancer agents[46]. Our recent study revealed a synergistic effect following the combination of lower concentrations of CIC with a bi-functional peptide which caused an increased toxicity against prostate cancer cells[47]. Further, in our studies, we demonstrated that EA, CIC, and piroctoneolamine (PO) exhibit potent selective toxicity on renal carcinoma cell lines by partly inhibiting the Wnt pathway[44]. We have also confirmed that the canonical Wnt signaling pathway is activated in Multiple myeloma through constitutively active β-catenin[48] and experimentally demonstrated a synergistic effect of a combination of thalidomide as well as lenalidomide and Wnt inhibitors (EA and CIC) in myeloma cells[5,23]. The Wnt inhibitors EA, CIC, PO and Griseofulvin (GF) were shown to reduce viability in murine and human pancreatic cancer cell lines[45].
As it resulted from a cell proliferation assay, viability was significantly decreased in all tested cancer cells and in a concentration - dependent manner. On a general note, lower concentrations of CIC produced greater killing effects on almost all cells compared to EA. Most interestingly, EA hardly altered the viability of our control cells CCD18Co, thus suggesting a favourable tolerability of EA. This was, however, different with CIC as it is a clinically used anti-fungal agent but showed a significant reduction in viability by 38% at 50 μM. The doses we used might have been too high as our results appear to be in accordance with several other reports showing that across multiple tumor types, CIC produced significant toxicity at low micro molar concentrations compared to EA[5,38]. Considering these facts, it may not be out of place to suggest that the degree of cell death perpetuated by CIC even at minimal concentrations across the cell types was not only due to β-catenin inhibition but a combination of several factors as described above. This is however subject to further investigation.
The summary of cell viabilities for EA at 200 μM (Fig 7B) shows that bladder and bronchial cancers were the least affected having a viability of 54.4% and 53.4% respectively. Previous studies have revealed that increased expression of dihydrodiol dehydrogenase (DDH), an enzyme previously implicated in the development of platinum resistance and described in an ethacrynic acid-resistant colon carcinoma cell line[49], was found to be a poor prognostic factor in patients with non-small cell lung cancer[50]. In their experiment, Rhodes and Twentyman discovered that the addition of EA as a potential modifier of melaphalan and cisplatin sensitivity in a drug resistant human lung cancer cell line A549 did not alter the dose response curves to these agents[51]. Furthermore, recently, Chen and his colleagues characterized the lung cancer cell line A549 as the most highly resistant cell line, which was defined as an adenocarcinoma / non-small cell carcinoma of the lung[52]. They reported that there is an up-regulation of certain proteins such as 14-3-3zeta and DDH, which are induced by stress-stimulation including chemo-preventive agents in A549 cell lines and these have been implicated in generation of drug resistance and correlated to poor treatment outcome. Thus, insensitivity of the A549 cell line to EA may be associated with over expression of DDH. Further experiments are required to certify this theory. However, bladder cancer was the most affected tumor type following treatment with CIC at 10 μM (Fig 7B). This result corresponds to that obtained by P. Rangarajan et al. during their in vitro study of the effect of CIC on the Non-Muscle Invasive Bladder Cancer (NMIBC) bladder cancer cell line T-24. Spheroid assay revealed that CIC inhibited the growth of T-24 at 4 μM by suppressing the stem cells[53].
Herein, we confirmed the anti-proliferative effects of CIC and EA and demonstrated their potent selective cytotoxicity on several cancer cell lines, which at least in part may be due to inhibition of Wnt/β-catenin signaling. The Wnt pathway is complex. Many of its components are shared with other pathways, generating cross - talks of varying intensities. Hence, it is sometimes difficult to clearly distinguish a direct influence of the drug on the Wnt pathway from its effects on the intersecting pathways. Therefore, for those cell lines that are significantly affected by the drugs, it is tempting to speculate that their enhanced sensitivity may be either related to the up regulation of such components as β-catenin, LEF-1 and its downstream effectors, even as we do not rule out the possibility of their potential influence on other signaling pathways. Not neglecting EA - mediated inhibition of NF-κB and MAPK signaling in various cell types[54], which may be a factor in its anti-tumor property besides a destabilization of the LEF-1/β-catenin transcription complex, we recommend that future studies should consider a validation of this report, chart the exact cross point interactions between the pathways and to what extent these action mechanisms synergistically compliment with the inhibition of the Wnt cascade. To this end, large data gene expression profiling experiments may be carried out with different dose paradigms of both drugs.
Conclusion
Our results are a confirmation of several other reports implicating the potential cytotoxic potency of EA and CIC on a wide array of tumor types. To a greater degree, from our analysis, colon cancer responded significantly to both drugs. Further, in vitro and in vivo research is recommended with special interest on colon cancer to provide deeper insight in the molecular mechanism and elucidate there in vivo efficacy for the treatment of various cancer diseases.
Acknowledgements: None
Conflicts of interest: The authors declare no conflict of interest.
References
1. Sato, T., Stange, D.E., Ferrante, M., et al. Long-term Expansion of Epithelial Organoids from Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. (2011) Gastroenterology 141(5): 1762–1772.
2. Zeng, Y.A., Nusse, R. Wnt Proteins Are Self-Renewal Factors for Mammary Stem Cells and Promote Their Long-Term Expansion in Culture. (2010) Cell Stem Cell 6(6): 568–577.
3. Blagodatski, A., Poteryaev, D., Katanaev, V.L. Targeting the Wnt pathways for therapies. (2014) Mol Cell Ther 2: 28.
4. Willert, K., Jones, K.A. Wnt signaling: is the party in the nucleus|| (2006) Genes Dev 20(11): 1394–1404.
5. Kim, Y., Schmidt, M., Endo, T., et al. Targeting the Wnt/beta-catenin pathway with the antifungal agent ciclopirox olamine in a murine myeloma model. (2011) In Vivo Athens Greece 25(6): 887–893.
6. Takahashi-Yanaga, F., Kahn, M. Targeting Wnt Signaling: Can We Safely Eradicate Cancer Stem Cells|| (2010) Clin Cancer Res 16(12): 3153–3162.
7. Shtutman, M., Zhurinsky, J., Simcha, I., et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. (1999) Proc Natl Acad Sci U S A 96(10): 5522–5527.
8. Chen, W., Chen, M., Barak, L.S. Development of small molecules targeting the Wnt pathway for the treatment of colon cancer: a high-throughput screening approach. (2010) Am J Physiol Gastrointest Liver Physiol 299(2): G293-300.
9. Prosperi, J.R., Goss, K.H. A Wnt-ow of opportunity: targeting the Wnt/beta-catenin pathway in breast cancer. (2010) Curr Drug Targets 11(9): 1074–1088.
10. Lu, D., Liu, J.X., Endo, T., et al. Ethacrynic Acid Exhibits Selective Toxicity to Chronic Lymphocytic Leukemia Cells by Inhibition of the Wnt/β-Catenin Pathway. (2009) PLoS ONE 4(12):e8294
11. Kim, Y., Alpmann, P., Blaum-Feder, S., et al. Increased in vivo efficacy of lenalidomide by addition of piroctone olamine. (2011) In Vivo Athens Greece 25(1): 99–103.
12. Schmeel, L.C., Schmeel, F.C., Kim, Y., et al. Flunarizine exhibits in vitro efficacy against lymphoma and multiple myeloma cells. (2015) Anticancer Res 35(3): 1369–1376.
13. Schmeel, L.C., Schmeel, F.C., Blaum-Feder, S., et al. In Vitro Efficacy of Naftifine against Lymphoma and Multiple Myeloma. (2015) Anticancer Res 35(11): 5921–5926.
14. Yao, H., Ashihara, E., Maekawa, T. Targeting the Wnt/β-catenin signaling pathway in human cancers. (2011) Expert Opin Ther Targets 15(7): 873–887.
15. Gong, Y., Bourhis, E., Chiu, C., et al. Wnt isoform-specific interactions with coreceptor specify inhibition or potentiation of signaling by LRP6 antibodies. (2010) PloS One 5(9): e12682.
16. Gurney, A., Axelrod, F., Bond, C.J., et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. (2012) Proc Natl Acad Sci U S A 109(29): 11717–11722.
17. Ciaccio, P.J., Shen, H., Jaiswal, A.K., et al. Modulation of detoxification gene expression in human colon HT29 cells by glutathione-S-transferase inhibitors. (1995) Mol Pharmacol 48(4): 639–647.
18. Aizawa, S., Ookawa, K., Kudo, T., et al. Characterization of cell death induced by ethacrynic acid in a human colon cancer cell line DLD-1 and suppression by N-acetyl-L-cysteine. (2003) Cancer Sci 94(10): 886–893.
19. Wang, R., Li, C., Song, D., et al. Ethacrynic acid butyl-ester induces apoptosis in leukemia cells through a hydrogen peroxide mediated pathway independent of glutathione S-transferase P1-1 inhibition. (2007) Cancer Res 67(16): 7856–7864.
20. Wu, W., Zhu, H., Fu, Y., et al. High LEF1 expression predicts adverse prognosis in chronic lymphocytic leukemia and may be targeted by ethacrynic acid. (2016) Oncotarget 7(16): 21631–21643.
21. Kim, Y., Gast, S-M., Endo, T., et al. In vivo efficacy of the diuretic agent ethacrynic acid against multiple myeloma. (2012) Leuk Res 36(5): 598–600.
22. Lacreta, F.P., Brennan, J.M., Nash, S.L., et al. Pharmakokinetics and bioavailability study of ethacrynic acid as a modulator of drug resistance in patients with cancer. (1994) J Pharmacol Exp Ther 270(3): 1186–1191.
23. Schmidt, M., Kim, Y., Gast, S-M., et al. Increased in vivo efficacy of lenalidomide and thalidomide by addition of ethacrynic acid. (2011) In Vivo Athens Greece 25(3): 325–333.
24. Zhao, G., Liu, C., Wang, R., et al. The synthesis of alpha,beta-unsaturated carbonyl derivatives with the ability to inhibit both glutathione S-transferase P1-1 activity and the proliferation of leukemia cells. (2007) Bioorg Med Chem 15(7): 2701–2707.
25. Jin, G., Lu, D., Yao, S., et al. Amide derivatives of ethacrynic acid: Synthesis and evaluation as antagonists of Wnt/β-catenin signaling and CLL cell survival. (2009) Bioorg Med Chem Lett 19(3): 606–609.
26. Furman, R.R., Asgary, Z., Mascarenhas, J.O., et al. Modulation of NF-kappa B activity and apoptosis in chronic lymphocytic leukemia B cells. (2000) J Immunol Baltim Md 1950 164(4): 2200–2206.
27. Horie, R., Watanabe, M., Okamura, T., et al. DHMEQ, a new NF-kappaB inhibitor, induces apoptosis and enhances fludarabine effects on chronic lymphocytic leukemia cells. (2006) Leukemia 20(5): 800–806.
28. Mook, R.A., Wang, J., Ren, X-R., et al. Structure-activity studies of Wnt/β-catenin inhibition in the Niclosamide chemotype: Identification of derivatives with improved drug exposure. (2015) Bioorg Med Chem 23(17): 5829–5838.
29. Zhou, H., Shen. T., Luo, Y., et al. The antitumor activity of the fungicide ciclopirox. (2010) Int J Cancer 127(10): 2467–2477.
30. Abrams, B.B., Hänel, H., Hoehler, T. Ciclopirox olamine: a hydroxypyridone antifungal agent. (1991) Clin Dermatol 9(4): 471–477.
31. Farinelli, S.E., Greene, L.A. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. (1996) J Neurosci 16(3): 1150–1162.
32. Leem, S-H., Park, J-E., Kim, I-S., et al. The possible mechanism of action of ciclopirox olamine in the yeast Saccharomyces cerevisiae. (2003) Mol Cells 15(1): 55–61.
33. Eberhard, Y., McDermott, S.P., Wang, X., et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. (2009) Blood 114(14): 3064–3073.
34. Wu, J., Liu, H., Zhang, G., et al. Antileukemia Effect of Ciclopirox Olamine Is Mediated by Downregulation of Intracellular Ferritin and Inhibition β-Catenin-c-Myc Signaling Pathway in Glucocorticoid Resistant T-ALL Cell Lines. (2016) PloS One 11(8): e0161509.
35. Torti, S.V., Torti, F.M. Iron and cancer: more ore to be mined. (2013) Nat Rev Cancer 13(5): 342–355.
36. Clement, P.M.J., Hanauske-Abel, H.M., Wolff, E.C., et al. The antifungal drug ciclopirox inhibits deoxyhypusine and proline hydroxylation, endothelial cell growth and angiogenesis in vitro. (2002) Int J Cancer 100(4): 491–498.
37. Song, S., Christova, T., Perusini, S., et al. Wnt inhibitor screen reveals iron dependence of β-catenin signaling in cancers. (2011) Cancer Res 71(24): 7628–7639.
38. Lee, N.P., Tsang, F.H., Shek, F.H., et al. Prognostic significance and therapeutic potential of eukaryotic translation initiation factor 5A (eIF5A) in hepatocellular carcinoma. (2010) Int J Cancer 127(4): 968–976.
39. Hoque, M., Hanauske-Abel, H.M., Palumbo, P., et al. Inhibition of HIV-1 gene expression by Ciclopirox and Deferiprone, drugs that prevent hypusination of eukaryotic initiation factor 5A. (2009) Retrovirology 6: 90.
40. Linden, T., Katschinski, D.M., Eckhardt, K., et al. The antimycotic ciclopirox olamine induces HIF-1alpha stability, VEGF expression, and angiogenesis. (2003) FASEB J 17(6): 761–763.
41. Shen, T., Huang, S. Repositioning the Old Fungicide Ciclopirox for New Medical Uses. (2016) Curr Pharm Des 22(28): 4443–4450.
42. Dihazi, G.H., Bibi, A., Jahn, O., et al. Impact of the antiproliferative agent ciclopirox olamine treatment on stem cells proteome. (2013) World J Stem Cells 5(1): 9–25.
43. Zhou, H., Shen, T., Shang, C., et al. Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway. (2014) Oncotarget 5(20): 10140–10150.
44. Schulz-Hausmann, S.A.V., Schmeel, L.C., Schmeel, F.C., et al. Targeting the Wnt/Beta-Catenin Pathway in Renal Cell Carcinoma. (2014) Anticancer Res 34(8): 4101–4108.
45. Wall, I., Schmidt-Wolf, I.G.H. Effect of Wnt inhibitors in pancreatic cancer. (2014) Anticancer Res 34(10): 5375–5380.
46. Koller, C.M., Kim, Y., Schmidt-Wolf, I.G.H. Targeting renal cancer with a combination of WNT inhibitors and a bi-functional peptide. (2013) Anticancer Res 33(6): 2435–2440.
47. Messina, C.S., Weiher, H., Schmidt-Wolf, I.G.H. Targeting Prostate Cancer with a Combination of WNT Inhibitors and a Bi-functional Peptide. (2017) Anticancer Res 37(2): 555–559.
48. Sukhdeo, K., Mani, M., Zhang, Y., et al. Targeting the β-catenin/TCF transcriptional complex in the treatment of multiple myeloma. (2007) Proc Natl Acad Sci 104(18): 7516–7521.
49. Ciaccio, P.J., Stuart, J.E., Tew, K.D. Overproduction of a 37.5-kDa cytosolic protein structurally related to prostaglandin F synthase in ethacrynic acid-resistant human colon cells. (1993) Mol Pharmacol 43(6): 845–853.
50. Chang, H-C., Chen, Y-L., Chan, C-P., et al. Overexpression of Dihydrodiol Dehydrogenase as a Prognostic Marker in Resected Gastric Cancer Patients. (2009) Dig Dis Sci 54(2): 342–347.
51. Rhodes, T., Twentyman, P.R. A study of ethacrynic acid as a potential modifier of melphalan and cisplatin sensitivity in human lung cancer parental and drug-resistant cell lines. (1992) Br J Cancer 65(5): 684–690.
52. Chen, J., Emara, N., Solomides, C., et al. Resistance to platinum-based chemotherapy in lung cancer cell lines. (2010) Cancer Chemother Pharmacol 66(6): 1103–1111.
53. Rangarajan, P., Ramalingam, S., Subramaniam, D, et al. Abstract 1895: Ciclopirox prodrug for the prevention and therapy of non-muscle invasive bladder cancer. (2015) Cancer Res 75(15): 1895–1895.
54. Han, Y., Englert, J.A., Delude, R.L., et al. Ethacrynic acid inhibits multiple steps in the NF-kappaB signaling pathway. (2005) Shock 23(1): 45–53.










