
In silico putative drug target identification in Enterobacter cloacae and homology modelling of a candidate drug target
Pramod Kumar Yadav*, Sudha Shukla, Sanjay Kumar Malhotra
Affiliation
In Silico Putative Drug Target Identification in Enterobacter Cloacae and Homology Modelling of a Candidate Drug Target
Corresponding Author
Yadav, P.K., et al. In Silico Putative Drug Target Identification in Enterobacter Cloacae and Homology Modelling of aCandidate Drug Target (2016) Bioinfo Proteom Img Anal 2(1): 65-70.
Citation
Yadav, P.K., et al. In Silico Putative Drug Target Identification in Enterobacter Cloacae and Homology Modelling of a Candidate Drug Target (2016) J Bioinfo Proteomics Rev 2(1): 1- 6.
Copy rights
© 2016 Yadav, P.K. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Enterobacter cloacae, KEGG, metabolic pathways, drug targets, homology modelling
Abstract
Enterobacter cloacae is a clinically significant Gram-negative, facultatively-anaerobic, rod-shaped bacterium, which has emerged as a prevalent nosocomial pathogen due to high level resistance to antimicrobial drugs. The availability of complete genome sequence of E. cloacae has paved the novel way to identify the new drug targets. In the present work comparative analysis of the metabolic pathways of the pathogen and human host was performed to identify the novel targets present in pathogen but absent in host. All enzymes involved in the metabolic pathways of E. cloacae were searched against the proteome of Homo sapiens using the BLASTp program using certain parametes. Approximately 44 unique non-homologous putative drug targets were identified. Among all these targets, 22 coding genes for putative targets were identified as essential in the DEG database for survival of the pathogen. After extensive literature survey, 8 targets were identified as potential drug. Among these targets UDP-N-acetylmuramate-L-alanine ligase (MurC) was chosen as candidate drug target for further study, because it was involved in many metabolic pathways of E. cloacae. The physico-chemical properties of the MurC protein were analyzed using the Protparam tool, and it was found the protein is stable in nature. The 3D structure of MurC enzyme was predicted using SWISS-MODEL and HHPred server respectively. Subsequently, all the predicted models were evaluated using the SAVES server. The best quality model was predicted by HHPred server which was having quality factor of 90.64, and approximately 90% of residues were falling in core regions of Ramachandran plot. The quality of the model was further validated by ProSA server. In future, the 3D structure of MurC in Enterobacter cloacae might be exploited for the discovery of novel inhibitors using structure-based drug design strategy that could potentially inhibit the pathogen.
Introduction
Enterobacter Cloacae is a clinically significant Gram-negative, facultatively-anaerobic, rod-shaped bacterium belonging to the family of Enterobacteriaceae. It has emerged as a prevalent nosocomial pathogen due to high level resistance to disinfectants and antimicrobial agents. The availability of complete genome sequence of E. cloacae has paved the new way to identify the novel drug targets. In the present work comparative analysis of the metabolic pathways of the pathogen and host was performed to identify the novel drug targets involved in pathogen but absent in Homo sapiens. All enzymes involved in the metabolic pathways of E. cloacae were searched against the proteome of H. sapiens using the BLASTp program. The threshold of percentage identity was set to as < 30%, E-value > 0.001, and the query coverage < 50. Using these parameters approximately 44 unique putative targets were identified. Out of those non-homologous targets, 22 coding genes for putative targets were identified as essential genes from the DEG database. Based on extensive literature search, 8 targets such as UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase, UDP-N-acetylmuramate-L-alanine ligase, UDP-N-acetylmuramoyl-L-alanyl-D-glutamate-2, 6-diaminopimelate ligase, UDP-N-acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase, UDP-N-acetylmuramate-L-alanine ligase (MurC) α-isopropylmalate synthase, UDP-N-Acetylglucosamine Enolpyruvyl Transferase, UDP-N-acetylenolpyruvylglucosamine reductase and Aspartate beta-semialdehyde dehydrogenase were identified as potential drug targets. Among these drug targets, UDP-N-acetylmuramate-L-alanine ligase (MurC) was chosen as potential therapeutic drug target. The homology modeling of MurC was performed using SWISS-MODEL and HHPred servers respectively. Subsequently, all the predicted models were evaluated using the SAVES server. The stereochemical parameters of all 3D models suggest that the best model was predicted by HHPred server. In order the refine the modeled structure, energy minimization was also performed using Deep View tool. The refined 3D structure was further validated by ProSA server. In future, the 3D structure of MurC in E. cloacae might be exploited for the discovery of novel inhibitors that could potentially inhibit this nosocomial pathogen.
------------------Full Text----------------------
Enterobacter Cloacae is a clinically significant Gram-negative, facultatively-anaerobic, rod-shaped bacterium belonging to the family of Enterobacteriaceae[1]. E. cloacae is a nosocomial pathogen that can cause a range of infections such as bacteremia, lower respiratory tract infection, skin and soft tissue infections, urinary tract infections, endocarditis, intra-abdominal infections, septic arthritis, osteomyelitis and ophthalmic infections[2]. ICU pathogens can cause morbidity and mortality and the management of these bacterial infections is complicated by the organism’s multiple antibody resistance. These bacteria contain beta-lactamase, which is undetectable in vitro and is highly resistant to antibiotics such as third generation cephalosporins. E. cloacae can be found on human skin and tissues as well as fruits, vegetables, and devices such as a hot water treatment tank. Although this organism is mainly a pathogen for human and causes disease. This pathogen has been used as a biological control for plant disease such as the seed-rotting oomycete in Pythium ultimum, and used to control insect pests on mulberry leaves and suppress disease[3]. This bacterium is an opportunistic type of bacteria which attacks (cause disease) in the host system (Homo sapiens) after it has been weakened by some other infections or injury. The E. cloacae is a type of species of Enterobacter, which is prevalent nosocomial pathogen due to high level resistance to disinfectants and antimicrobial agents[4]. The E. cloacae ATCC 13047 strain was first isolated from human cerebrospinal fluid by Edwin Oakes Jordan in 1890 and is the type strain of E. cloacae subsp. cloacae[5]. The aim of present research work was to identify potential therapeutic drug targets in E. cloacae using metabolic pathway using metabolic pathways analysis, and to model the 3D structure of candidate drug target.
Materials and Methods
Drug target identification In the present work KEGG pathway database has been used (https://www.genome.jp/kegg/) as a source of metabolic pathway information about E. cloacae strain ATCC 13047. Total 86 different types of metabolic pathways were analyzed, and the enzymes involved in these unique pathways were identified from KEGG database[6]. The protein sequences of all the enzymes involved in different metabolic pathways of E. cloacae were subjected to BLASTp[7] search against the proteome of Homo sapiens. In order to identify the non-homologous encoding genes to the host (human), the threshold values were set to as E-value > 0.001, % identity < 30 %, and query coverage < 50%. Using these parameters, 172 targets have been identified as non-homologous to the H. sapiens. Further analysis of the identified putative targets was carried out, and the enzymes present in multiple pathways (duplicates) of the pathogen were removed. Finally, 44 targets were identified as unique putative drug targets in E. cloacae. The genes encoding for the important enzymes have been further searched in the DEG database (https://tubic.tju.edu.cn/deg/)[8] to identify the essentiality and non-essentiality of the genes for the survival of the pathogen. Among 44 drug targets, 22 targets were identified as the essential for the survival of E.cloacae. Out of these 22 targets, 8 drug targets were identified as potential therapeutic drug target from the extensive literature review. Among these targets, MurC was selected as candidate drug target for further study.
Sub-cellular localization of putative drug targets: The sub-cellular localization (SCL) of all 22 essential gene products in E. cloacae was predicted using the PSORTb v3.0.2 program (https://www.psort.org/psortb/). It is the first sub-cellular localization predictor exclusively devised for all prokaryotes, including archaea and bacteria with atypical membrane/cell wall topologies. It handles archaeal sequences as well as Gram-positive and Gram-negative bacterial protein sequences. This program consists of various analytical modules, each of which analyzes one biological feature known to influence or be characteristic of sub-cellular localization. The modules may act as a binary predictor, classifying a protein as either belonging or not belonging to a particular localization site, or they may be multi-category, able to assign a protein to one of several localization sites.
Protein-protein interaction network analysis: In order to analyze the molecular interaction networks of candidate drug target UDP-N-acetylmuramate-L-alanine ligase (MurC), the protein-protein interaction study was carried out from the architecture of E. cloacae interactome using the STITCH v3.1web server (https://stitch.embl.de/). This server explores known and predicted interactions of proteins and chemicals.
Physico-chemical propertics prediction of MurC: The ProtParam computes various physico-chemical properties that can be deduced from a protein sequence. Various parameters include the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). These physico-chemical properties of MurC protein in E. cloacae were analyzed using the ProtParam server (https://web.expasy.org/protparam/).
Homology Modeling and evaluation of MurC: The homology modeling was performed to build a three-dimensional model of UDP-N-acetylmuramate-L-alanine ligase (MurC) based on one more related protein of known structure (the template). The protein sequence of MurC was retrieved from the Uniprot database and submitted to Swiss-model and HHPred server respectively. The PDB file of the modeled structure was downloaded and 3D structures of modeled structures were visualized using the Rasmol tool. Subsequently, the Structure Analysis and Verification Servers (SAVES) were used to evaluate the stereo-chemical properties of all the models predicted by various servers. SAVES uses PROCHECK, WHAT_IF, ERRAT, VERIFY 3D, and PROVE molecules to check any anomaly present in the structure. The best predicted model of MurC was of HHpred server. The energy minimization of the modeled structure of MurC was carried out to improve the overall quality of the model using the DeepView tool. The energy minimized modelled structure of MurC was further validated using SAVES and ProSA server respectively.
Results and Discussion
Drug target identification
The availability of complete genome sequence has paved the new way to identify the novel potential drug targets in E. cloacae. Earlier several potential drug targets have been identified in numerous pathogens using the metabolic pathways analysis[9-11]. In the present work, metabolic pathways of E. cloacae were analyzed, and total 86 metabolic pathways were present in the KEGG pathways databases. All the enzymes involved in the metabolic pathways of E. cloacae were searched against the proteome of H. sapiens using the BLASTp program. The threshold of percentage identity was set to as < 30%, E-value > 0.001, and the query coverage < 50 %. Total 172 putative drug targets were identified. Out of 172 drug targets, there was found some targets or genes which were involved in more than one metabolic pathway. All the duplicates targets were removed from the list, and total 44 unique putative drug targets have been found. The genes encoded for 44 unique putative drug targets were again searched against the DEG (Database of Essential Genes) database to identify the essentiality of the genes for the survival of E. cloacae. Total 22 encoding genes have been identified as essential for the survival of this pathogen (Table 1).
Table 1: List of Essential genes for Enterobacter Cloacae
| S.No | Gene ID | Gene Name | DEG ID | TARGETS | Pathways |
| 1 | ECL_00191 | dlgD | DEG10050373 | 2,3-diketo-L-gulonate reductase[EC:1.1.1.130] | Pentose and glucuronate interconversions |
| Ascorbate and aldarate metabolism | |||||
| 2 | ECL_05074 | rhaD | DEG10040567 | rhamnulose-1-phosphate aldolase [EC:4.1.2.19] | Pentose and glucuronate interconversions |
| Fructose and mannose metabolism | |||||
| 3 | ECL_00002 | dnaN | DEG10040692 | DNA polymerase III subunit beta [EC:2.7.7.7] | Purine metabolism |
| Pyrimidine metabolism | |||||
| 4 | ECL_03056 | dnaN | DEG10110044 | DNA polymerase III subunit delta [EC:2.7.7.7] | Purine metabolism |
| Pyrimidine metabolism | |||||
| 5 | ECL_04389 | ureC | DEG10100307 | urease subunit alpha [EC:3.5.1.5] | Purine metabolism |
| Arginine and proline metabolism | |||||
| 6 | ECL_04795 | asd | DEG10040525 | aspartate-semialdehyde dehydrogenase [EC:1.2.1.11] | Glycine, serine and threonine metabolism |
| Cysteine and methionine metabolism | |||||
| Lysine biosynthesis | |||||
| 7 | ECL_02168 | asd | DEG10130082 | aspartate-semialdehyde dehydrogenase [EC:1.2.1.11] | Glycine, serine and threonine metabolism |
| Cysteine and methionine metabolism | |||||
| Lysine biosynthesis | |||||
| 8 | ECL_04130 | tdcG | DEG10040468 | L-serine dehydratase [EC:4.3.1.17] | Glycine, serine and threonine metabolism |
| Cysteine and methionine metabolism | |||||
| 9 | ECL_02018 | metE | N/A* | 5-methyltetrahydropteroyl triglutamate--homocysteine methyltransferase [EC:2.1.1.14] | Cysteine and methionine metabolism |
| Selenocompound metabolism | |||||
| 10 | ECL_04966 | metE | DEG10100167 | 5-methyltetrahydropteroyl triglutamate--homocysteine methyltransferase [EC:2.1.1.14] | Cysteine and methionine metabolism |
| Selenocompound metabolism | |||||
| 11 | ECL_04024 | leuA | DEG10100584 | 2-isopropylmalate synthase [EC:2.3.3.13] | Valine, leucine and isoleucine biosynthesis |
| Glycerophospholipid metabolism | |||||
| 12 | ECL_00882 | murE | DEG10040016 | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate--2,6-diaminopimelate ligase [EC:6.3.2.13] | Lysine biosynthesis |
| Peptidoglycan biosynthesis | |||||
| 13 | ECL_00883 | murF | DEG10040617 | UDP-N-acetylmuramoyl-tripeptide--D-alanyl-D-alanine ligase [EC:6.3.2.10] | Lysine biosynthesis |
| Peptidoglycan biosynthesis | |||||
| 14 | ECL_02394 | ARO4 | DEG10040290 | 3-deoxy-7-phosphoheptulonate synthase [EC:2.5.1.54] | Tryptophan metabolism |
| Phenylalanine, tyrosine and tryptophan biosynthesis | |||||
| 15 | ECL_02983 | aroG-2 | DEG10040290 | 3-deoxy-7-phosphoheptulonate synthase [EC:2.5.1.54] | Tryptophan metabolism |
| Phenylalanine, tyrosine and tryptophan biosynthesis | |||||
| 16 | ECL_03678 | aroC | DEG10050092 | chorismate synthase [EC:4.2.3.5] | Tryptophan metabolism |
| Phenylalanine, tyrosine and tryptophan biosynthesis | |||||
| 17 | ECL_00885 | murD | DEG10110007 | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase [EC:6.3.2.9] | D-Glutamine and D-glutamate metabolism |
| Peptidoglycan biosynthesis | |||||
| 18 | ECL_00888 | murC | DEG10040020 | UDP-N-acetylmuramate—L-alanine ligase [EC:6.3.2.8] | D-Glutamine and D-glutamate metabolism |
| Peptidoglycan biosynthesis | |||||
| 19 | ECL_00944 | panC | N/A* | pantoate--beta-alanine ligase [EC:6.3.2.1] | beta-Alanine metabolism |
| Pantothenate and CoA biosynthesis | |||||
| 20 | ECL_04101 | cysD | DEG10130159 | sulfate adenylyltransferase subunit 2 [EC:2.7.7.4] | Selenocompound metabolism |
| Sulfur metabolism | |||||
| 21 | ECL_04571 | murA | DEG10040662 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase [EC:2.5.1.7] | Amino sugar & Nt. sugar metabolism |
| Peptidoglycan biosynthesis | |||||
| 22 | ECL_04946 | murB | DEG10110210 | UDP-N-acetylenolpyruvylglucosamine reductase [EC:1.3.1.98] | Amino sugar & Nt. sugar metabolism |
| Peptidoglycan biosynthesis |
On the basis of extensive literature review, out of 22 Essential drug targets, 8 putative drug targets were reported as potential therapeutic drug targets in various pathogens. These targets were also involved in more than one metabolic pathway of E. cloacae. Those potential drug targets are listed below:
- i. UDP-N-acetylmuramoylalanyl-D-glutamate-2, 6-diaminopimelate ligase
- ii. UDP-N-acetylmuramoyl-tripeptide--D-alanyl-D-alanine ligase
- iii. UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase
- iv. UDP-N-acetylmuramate-L-alanine ligase
- v. α-Isopropylmalate synthase
- vi. UDP-N-Acetylglucosamine Enolpyruvyl Transferase
- vii. UDP-N-acetylenolpyruvylglucosamine reductase
- viii. Aspartate-beta-semialdehyde dehydrogenase
Sub-cellular localization of putative drug targets in E. cloacae In order to predict the sub-cellular localization (SCL) of all 22 gene products (putative drug targets) in E. cloacae, the PSORTb v3.0.2 program was used. In this program several modules were used for the prediction of sub-cellular localization of bacterial proteins. All 22 unique drug targets crucial for the survival of E. cloacae were subjected for the prediction of sub-cellular localization. It was found that all the targets were localized in cytoplasmic region (Appendix Table I), which might be easily accessible in bacterial cell being targeted for inhibitor designing.
Appendix Table I: Sub-cellular localization of putative drug targets in E.cloacae
| S.N. | Gene ID | Gene Name | DEG ID TARGETS | Sub-cellular localization | Localization probability score |
| 1 | ECL_00191 | dlgD | 2,3-diketo-L-gulonate reductase[EC:1.1.1.130] | cytoplasmic | 9.97 |
| 2 | ECL_05074 | rhaD | rhamnulose-1-phosphate aldolase [EC:4.1.2.19] | cytoplasmic | 9.97 |
| 3 | ECL_00002 | dnaN | DNA polymerase III subunit beta [EC:2.7.7.7] | cytoplasmic | 9.97 |
| 4 | ECL_03056 | dnaN | DNA polymerase III subunit delta [EC:2.7.7.7] | cytoplasmic | 9.97 |
| 5 | ECL_04389 | ureC | urease subunit alpha [EC:3.5.1.5] | cytoplasmic | 9.97 |
| 6 | ECL_04795 | asd | aspartate-semialdehyde dehydrogenase [EC:1.2.1.11] | cytoplasmic | 9.97 |
| 7 | ECL_02168 | asd | aspartate-semialdehyde dehydrogenase [EC:1.2.1.11] | cytoplasmic | 9.97 |
| 8 | ECL_04130 | tdcG | L-serine dehydratase [EC:4.3.1.17] | cytoplasmic | 9.97 |
| 9 | ECL_02018 | metE | 5-methyltetrahydropteroyl triglutamate--homocysteine methyltransferase [EC:2.1.1.14] | cytoplasmic | 9.97 |
| 10 | ECL_04966 | metE | 5-methyltetrahydropteroyl triglutamate--homocysteine methyltransferase [EC:2.1.1.14] | cytoplasmic | 9.97 |
| 11 | ECL_04024 | leuA | 2-isopropylmalate synthase [EC:2.3.3.13] | cytoplasmic | 9.97 |
| 12 | ECL_00882 | murE | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate--2,6-diaminopimelate ligase [EC:6.3.2.13] | cytoplasmic | 9.97 |
| 13 | ECL_00883 | murF | UDP-N-acetylmuramoyl-tripeptide--D-alanyl-D-alanine ligase [EC:6.3.2.10] | cytoplasmic | 9.97 |
| 14 | ECL_02394 | ARO4 | 3-deoxy-7-phosphoheptulonate synthase [EC:2.5.1.54] | cytoplasmic | 9.97 |
| 15 | ECL_02983 | aroG-2 | 3-deoxy-7-phosphoheptulonate synthase [EC:2.5.1.54] | cytoplasmic | 9.97 |
| 16 | ECL_03678 | aroC | chorismate synthase [EC:4.2.3.5] | cytoplasmic | 9.97 |
| 17 | ECL_00885 | murD | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase [EC:6.3.2.9] | cytoplasmic | 9.97 |
| 18 | ECL_00888 | murC | UDP-N-acetylmuramate—L-alanine ligase [EC:6.3.2.8] | cytoplasmic | 9.97 |
| 19 | ECL_00944 | panC | pantoate--beta-alanine ligase [EC:6.3.2.1] | cytoplasmic | 9.97 |
| 20 | ECL_04101 | cysD | sulfate adenylyltransferase subunit 2 [EC:2.7.7.4] | cytoplasmic | 9.97 |
| 21 | ECL_04571 | murA | UDP-N-acetylglucosamine 1-carboxyvinyltransferase [EC:2.5.1.7] | cytoplasmic | 9.97 |
| 22 | ECL_04946 | murB | UDP-N-acetylenolpyruvylglucosamine reductase [EC:1.3.1.98] | cytoplasmic | 9.97 |
Since the UDP-N-acetylmuramate-L-alanine ligase (MurC) was involved in multiple pathways, and it is also essential for the survival of E. cloacae, it was selected as a candidate drug target for further analysis.
Protein-protein interaction network analysis
The protein-protein interactions (PPIs) are crucial in almost all biological processes occur in any organism. Using global protein interaction network (interactome) analysis, the relationships between genes/proteins can be understood in an effective way. The sub-network analysis and hub prioritization of drug target MurC in E. cloacae interactome was carried out using the STITCH 3.1 server.
After analyzing the protein-protein interaction sub-network of MurC in E. cloacae interactome, it was observed that this target is a crucial hub which is highly connected with murD, murB, Ent638_0638, murG, UDP-N-acet.ani, murF, murE, Ent638_0635, mraY, and Ent638_0639 proteins (Figure1). The protein-protein interaction of FBA was predicted with > 0.9 confidence score (Approximate probability), which shows strong association with these proteins (Table 2).
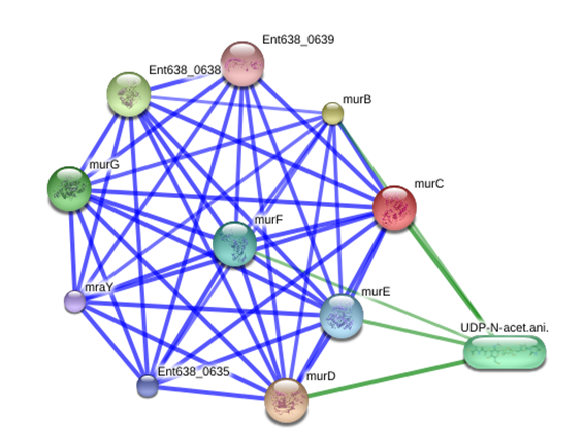
Figure 1: Protein interaction network of UDP-N-acetylmuramate--L-alanine ligase (MurC) in E. cloacae (Protein-protein interactions are shown in blue, chemical-protein interactions in green and interactions between chemicals in red. Stronger associations are represented by thicker lines)
Table 2: Predicted functional partners of murC protein in the proteome of E. cloacae
| S. No. | Protein/chemical code | Predicted associated protein/chemical in the network | Score (Probability score) |
| 1 | murD | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase | 0.999 |
| 2 | murB | UDP-N-acetylenolpyruvoylglucosamine reductase | 0.997 |
| 3 | Ent638_0638 | D-alanine--D-alanine ligase | 0.996 |
| 4 | murG | undecaprenyldiphospho-muramoylpentapeptide beta-N- acetylglucosaminyltransferase | 0.993 |
| 5 | UDP-N-acet.ani | UDP-N-acetylmuramoyl-L-alanine | 0.992 |
| 6 | murF | UDP-N-acetylmuramoyl-tripeptide--D-alanyl-D- alanine ligase | 0.989 |
| 7 | murE | UDP-N-acetylmuramoylalanyl-D-glutamate--2, 6-diaminopimelate ligase | 0.988 |
| 8 | Ent638_0635 | cell division protein FtsW | 0.984 |
| 9 | mraY | phospho-N-acetylmuramoyl-pentapeptide- transferase | 0.981 |
| 10 | Ent638_0639 | cell division protein FtsQ | 0.981 |
Physico-chemical propertics prediction of MurC
Various physico-chemical parameters such as the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity (GRAVY), were used to analyze the MurC protein of E. cloacae by ProtParam server[12]. (Table II) (Appendix) depicts that total 491 were present in the protein, in which the percentage of Alanine was the highest (11.4) and the percentage of Tryptophan was the lowest (0.2). It also showed that the MurC is a stable protein with an instability index of 30.73. According to this program, a protein whose instability index is smaller than 40 is predicted as stable; on the other hand, a value above 40 predicts that the protein maybe unstable. Above value suggest that this protein is stable and catalytically active under room temperature. The MurC protein has a theoretical pI value of 5.61, and its molecular weight was found to be 53382.5, which suggest moderate size of protein.
Appendix Table II: Physico-chemical properties of MurC protein
| Amino acids and their properties | Percentage (%)/Values |
| Ala (A) | 11.4 |
| Asn (N) | 3.7 |
| Cys (C) | 0.4 |
| Glu (E) | 5.9 |
| His (H) | 3.9 |
| Leu (L) | 8.8 |
| Met (M) | 2.4 |
| Pro (P) | 4.7 |
| Thr (T) | 5.9 |
| Tyr (Y) | 2.2 |
| Pyl (O) | 0.0 |
| Arg (R) | 6.5 |
| Asp (D) | 6.5 |
| Gln (Q) | 3.9 |
| Gly (G) | 8.4 |
| Ile (I) | 6.7 |
| Lys (K) | 2.6 |
| Phe (F) | 3.7 |
| Ser (S) | 4.5 |
| Trp (W) | 0.2 |
| Val (V) | 7.7 |
| Sec (U) | 0.0 |
| Number of amino acids | 491 |
| Molecular weight | 53382.5 |
| Theoretical pI | 5.61 |
| Instability index | 30.73(stable) |
| Aliphatic index | 94.22 |
| Grand average of hydropathicity (GRAVY) | -0.111 |
| Instability index | 30.73(stable) |
Homology Modeling and evaluation of MurC
The homology modeling was performed to build a three-dimensions (3D) structure of candidate drug target UDP-N-acetylmuramate-L-alanine ligase (MurC) based on one more related protein of known structure (the template). The protein sequence of MurC was retrieved from the Uniprot database and submitted to Swiss-model and HHPred server respectively. The 3D structure of target was predicted using the template MurC from Yersinia pestis (PDB ID: 4hv4), which was having 85% sequence identity and 5.3e-90 E-value. The higher percentage of sequence identity and lower E-value suggest that the template is closely related to the target protein, and better quality 3D structure can be predicted using the homology modeling method. The PDB files[13,14] of the modeled structure was downloaded and visualized using the Rasmol tool. Subsequent to structure prediction, Structure Analysis and Verification Server (SAVES)[15-17] was used to evaluate the stereo-chemical properties of all the models predicted by various servers. SAVES uses PROCHECK, WHAT_IF, ERRAT, VERIFY 3D, and PROVE molecules to check any anomaly present in the structure. The best predicted model of MurC was of HHpred server. The energy minimization of the modeled structure of MurC was done to improve the quality of the model using SPDBV tool[18]. The energy minimised of modelled structure of MurC was further validated using SAVES and ProSA server respectively.
The model predicted by HHpred server shown that on Ramachandran plot, 91.8% amino acid residue were falling in core, 6.8% allowed, 0.7% generously allowed and 0.7% disallowed regions and bad contacts were 12. Verify 3D result shows that 91.87% of residues had an average 3D-1D score>0.2, the overall quality factor of the model was 85.443 (Figure 2).
In order to improve the overall quality of the predicted model of MurC, the energy minimization was carried out 3 times iteratively using the GROMOS96 force field implemented in the Deep View tool Through this process the molecular structure was brought to its minimum energy conformation and bad contacts were also reduced significantly from 5 to 1 (Table 3 ).
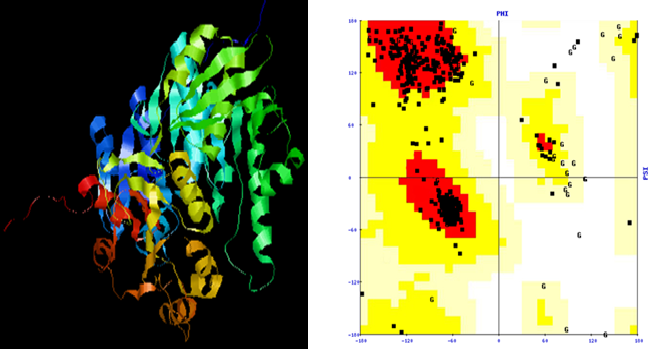
Figure 2: 3D structure and Ramachandran plot of MurC
Table 3: Evaluation of 3D structure of MurC after Energy minimization
| Number of times energy minimization | Procheck result (Ramachandran plot) | Verify 3D result (% of the residues had an average 3D-1D score >0.2 | Errat (overall quality factor) | Bad Contacts |
| 1 | 90.8% core 7.5% Allowed 0.9% gener 0.7% disallowed | 92.68% | 89.149 | 5 |
| 2 | 90.8% core 7.5% Allowed 0.9% gener 0.7% disallowed | 92.68% | 90.426 | 3 |
| 3 | 89.4% core 8.9% allowed 1.2% gener 0.5% disallowed | 92.68% | 90.638 | 1 |
ProSA (Protein structure analysis)
The modeled 3D structure of UDP-N-acetylmuramate-L-alanine ligase (MurC) was revalidated by the ProSA server[19]. (Figure 3) shows the Z-Score, which indicates the overall quality of the modeled structure.
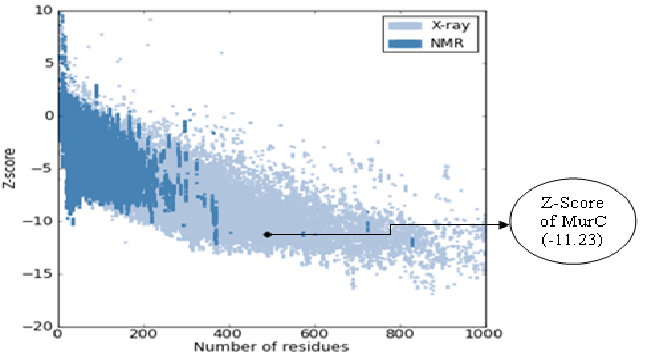
Figure 3: Overall quality of model predicted by ProSA server
The Z-Score of MurC protein was found to be -11.23, which indicates good quality of protein. The dark blue colour indicates NMR whereas light blue indicates X-ray technique region. Here the Z-Score lies in the X-ray plot region which signifies that MurC is within the range of scores typically found for native proteins of similar size.
The validation results show that the predicted 3D structure of UDP-N-acetylmuramate-L-alanine ligase (MurC) is of better quality. A good quality model would be expected to have more than 90% amino acid residues in the most favored (core) regions, and the results suggest that the predicted model is of good quality. The modeled structure of MurC can be exploited as potential therapeutic drug target using structure-based drug designing strategy against the E. cloacae.
Conclusion
The metabolic pathways in the genome of E. cloacae were analyzed, and 8 putataive targets were identified as potential drug targets. Among these targets, UDP-N-acetylmuramate-L-alanine ligase (MurC) was chosen as potential therapeutic drug target. The protein-protein interaction sub-network of MurC suggests that this target is a crucial hub, which is highly connected with several essential proteins. The 3D structure of MurC protein was predicted using the homology modeling method. After stereo-chemical evaluation and further refinement, it was found that predicted model is of good quality. In future, the 3D structure of MurC in E. cloacae might be exploited for the discovery of novel inhibitors that could potentially inhibit the pathogen.
References
- 1. Nishijima, K. A. (1999). Enterobacter cloacae Crop Knowledge Master
- 2. Fraser and Susan, L. (2007). Enterobacter infections. Emedicine.
- 3. Dijk, K.V., Nelson, E.B. Fatty Acid Competition as a Mechanism by which Enterobacter cloacae Suppresses Pythium ultimum sporangium germination and damping-off. (2000) Appl Environ Microbiol 66(12): 5340-5347.
- 4. John, J.F., Sharbaugh, R.J., Bannister, E.R. Enterobacter cloacae: bacteremia, epidemiology, and antibiotic resistance. (1982) Rev Infect Dis 4(1): 13-28.
- 5. Hoffmann, H., Stindl, S., Ludwig, W., et al. Reassignment of enterobacter dissolvens to Enterobacter cloacae as E. cloacae subspecies dissolvens comb. nov. and emended description of Enterobacter asburiae and Enterobacter kobei. (2005) Syst Appl Microbiol 28(3): 196-205.
- 6. Kanehisa, M., Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. (2000) Nucleic Acids Res 28(1): 27-30.
- 7. Altschul, S.F., Gish, W., Miller, W., et al. Basic local alignment search tool. (1990) J Mol Biol 215(3): 403-410.
- 8. Zhang, R., Lin, Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. (2009) Nucleic Acids Res 37: D455-D458.
- 9. Singh, S., Singh, G., Sagar, N., et al. Insight into trichomonas vaginalis genome evolution through metabolic pathways comparison. (2012) Bioinformation. 8(4): 189-195.
- 10. Yadav, P.K., Singh, G., Singh, S., et al. Potential therapeutic drug target identification in Community Acquired-Methicillin Resistant Staphylococcus aureus (CA-MRSA) using computational analysis. (2012) Bioinformation 8(14): 664-672.
- 11. Yadav, P.K., Pandey, H.K. In silico drug target identification in Legionella pneumophila strain Paris: modelling and molecular dynamics simulation of a candidate enzyme phosphoglyceromutase (PGM). (2013) Int J Comput Bioinfo In Silico Model 2(3): 132-137.
- 12. Gasteiger, E., Hoogland, C., Gattiker, A., et al. Protein Identification and Analysis Tools on the ExPASy Server. (2005) The Proteomics Protocols Handbook 571-607.
- 13. Berman, H.M. The Protein Data Bank: a historical perspective. (2008) Acta Crystallogr A 64(1): 88-95.
- 14. Meyer, E.F. The first years of the Protein Data Bank. (1997) Protein Sci 6(7): 1591-1597.
- 15. Laskowski, R.A., MacArthur, M.W., Moss, D.S. et al. PROCHECK: A program to check the stereochemical quality of protein structures. (1993) J Appl Cryst 26: 283-291.
- 16. Vriend, G. WHAT IF: A molecular modeling and drug design program. (1990) J Mol Graph 8(1): 52-56.
- 17. Luthy, R., Bowie, J.U., Eisenberg, D. Assessment of protein models with three-dimensional profiles. (1992) Nature 356(6364): 83-85.
- 18. Guex, N., Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. (1997) Electrophoresis 18(15): 2714-2723.
- 19. Wiederstein, M., Sippl, M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. (2007) Nucleic Acids Res 35: W407-W410.










