
Interpreting Large-Scale Phylogenetic Models of Mammal Class Diversification Based on Mitochondrial Biomarkers
Affiliation
Pangaea Biosciences, Department of Research & Development, Miami, FL USA
Corresponding Author
Tommy Rodriguez, Pangaea Biosciences, Department of Research & Development, Miami, FL USA, E-mail: trodriguez@pangaeabio.com / bio_nerd@outlook.com
Citation
Rodriguez,T. Interpreting Large-Scale Phylogenetic Models of Mammal Class Diversification Based on Mitochondrial Biomarkers (2017) Bioinfo Proteom Img Anal 2(2): 162- 168.
Copy rights
© 2017 Rodriguez,T. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
mammal evolution, Kailgn, mtDNA, Bayesian models, molecular phylogenetics
Abstract
This investigation reexamines mammal class diversification from a comparative phylogenetic perspective. By reconstructing a large-scale phylogeny based on mitochondrial biomarkers, I seek to reaffirm models of mammal class diversification among lineages that endured through the K-T event and onward. Two hundred thirty-five complete mtDNA sequences and sixty-two major taxa families within the class mammalia were represented in this study. My results will show that divergence among early eutherian mammals can be traced to a single modern group that shares homologous traits with the oldest eutherian fossil species ever found. Moreover, molecular clock analysis of phylogenetic reconstruction highlights the position taken by others rapid diversification between marsupial and placental mammals may have occurred much earlier than traditional models presume.
Introduction
The great diversity of Cenozoic mammalian families includes some five thousand four hundred documented species of living mammals assigned to twenty-six orders Vié et al., 2007[1]. Yet, despite the abundant amount of diversity, much remains unknown about their ancestral lineages prior to the Paleocene epoch. Several mammalian orders are only interpreted as diversifying immediately after the K–Pg boundary, including the largest animals on the planet, the great whales, as well as some of the most intelligent, such as elephants, primates and cetaceans Vaughan et al., 2013[2]. In particular, it is thought that modern mammals arose in the Paleogene and Neogene periods of the Cenozoic, after the extinction of non-avian dinosaurs Robertson et al., 2004[3]. Prior to the K–Pg boundary, mammalian species are generally found small, comparable in size to rats; this small size would have helped them find shelter in protected environments Robertson et al., 2004[3].
When most of the non-avian dinosaurs perished, the surviving mammals diversified into the dinosaurs’ niches, where they remain today Luo et al., 2011[4]. Interestingly, recent investigations in phylogeny research have reported two significant findings: (a) rapid diversification among primitive eutherian mammals may have occurred much earlier than traditional models presume Meredith et al., 2011[5] & Eizirik et al., 2004[6]; and (b) the oldest living lineages of modern eutherians can be traceable to a single modern group Brandt et al., 2016[7]. A number of phylogenetic studies involving mitochondrial biomarkers have successfully reconstructed the phylogeny of mammals at different levels and scales of the taxonomic hierarchy in order to provide a basis for standardizing methodologies Tobe et al., 2010[8]. One such paper published in March 2016, postulates that a single group of modern mammals predates the extinction of the dinosaurs Brandt et al., 2016[7]. Brandt et al.2016[7] used two methods to sequence the traces of mitochondrial DNA collected, and confirmed that it diverged from all other mammals approximately 78 Myr. Mitochondrial DNA provides the ideal test case for these kinds of analysis, because all genes are inherited as a single unit and thus have a single evolutionary history Corneli et al., 2000[9].
Genetic analysis has played an increasingly important role in confirming existing or establishing radically different mammalian groupings and phylogenies Tobe et al., 2010[8]. A combinational approach may too provide an ideal method for verification, where genetic analysis is crossed-referenced with morphological models. As it related to this study, in 2011 researchers reported on the discovery of a fossil mammal found in China that would have lived alongside the dinosaurs and that, at 160 million years old, represents one of the earliest mammals known today (Luo et al., 2011[4]. Juramaia sinensis, a furry rodent- like animal just a few centimeters long, is thought to be the oldest known common ancestor of modern placentals, or a very closely related cousin to that common ancestor Luo et al., 2011[4]. The correlation between Juramaia sinensis and the findings present here will be further highlighted in later sections.
Fossils alone are not always available and sufficiently informative, and phylogenetic methods based on models of character evolution can be unsatisfactory Figuet et al., 2014[10]. Genomic data offers a more robust opportunity to estimate these ancestral lineages Figuet et al.,2014[10]. This investigation features several techniques in bioinformatics for phylogeny research. By reconstructing a large-scale phylogeny based on mitochondrial biomarkers, I seek to reaffirm models of mammal class diversification among lineages that endured through the K-T event and onward. Two hundred thirty-five complete mtDNA sequences and sixty-two major taxa families within the class mammalia were represented in this study. My primary objective is to outline a practical framework by which time-extended lineages could be assessed and evaluated, in order to better understand the evolutionary trajectories that led to the abundant diversity within mammalian families.
Sixty-two taxa families for sequence selection
Mitochondrial biomarkers serve a practical use in large-scale processing for comparative sequence analysis. As such, mitochondrial sequences have the advantage of being translatable, and at the level of species and genera usually do not contain high numbers of length-variable regions Bohle et al., 2012[11]. Two key factors support the application of mtDNA for molecular phylogenetics: (a) technically, complete mtDNA datasets are light-weight compared to larger genomic datasets, which can create systematic bottlenecks during processing and execution that lead to erroneous inferences; and (b) mitochondrial DNA accumulates nucleotide substitutions relatively rapidly, due to lack of repair mechanisms that slow down the molecular clock Brown, 2002[12]. In theory, this feature makes complete mtDNA sequences suitable for inquiries involving species-level and genus-level classifications.
Here, the mtDNA sequence selections relied exclusively on preexisting order classifications, as to represent the major family groups within the class mammalia. The NCBI nucleotide databank was the platform where each mtDNA sequence was collected. On a number of different instances, BLAST similarity searches were required to identify the most homologous sequence candidates among the many mammalian groups selected for purposes of this study. And, species selection was appropriated on the basis of cross-referencing independent data with respect to morphology alone. From these findings, several distinct FASTA data files containing a combination of two hundred thirty-five mtDNA sequences that represent the sixty-two major families were compiled. Each individual data file was later combined into a master file that contains all the family types outlined below. It should then be noted, that neither file exceeded 4,000 KB. See additional notes for accession numbers in Arnold, M.L., 1997[13].
Multiple Sequence Alignment
A common theme in past studies often involves utilizing Kalign for Multiple Sequence Alignment (MSA) alongside PHYLIP neighbor-joining method. This technique has proven particularly effective. An accurate and fast MSA algorithm, Kalign is a dependable algorithmic selection for purposes of obtaining highly-robust base-pair alignments Lassmann & Sonnhammer[14]. Kalign is an extension of Wu-Manber approximate pattern-matching algorithm, based on Levenshtein distances. This strategy enables Kalign to estimate sequence distances faster and more accurately than other popular iterative methods. Lassmann and Sonnhammer show that Kalign is about 10 times faster than ClustalW and, depending on the alignment size, up to 50 times faster than other iterative methods; Kalign also delivers better overall resolution Lassmann & Sonnhammer, 2005 [14].
Runtime execution directly affects output resolution. Moreover, eliminating or reducing system bottlenecks is one prime objective of algorithmic selection for MSA, as it helps narrow the scope for error. Comparisons done between three MSA algorithms (Kalign, ClustalW & MAFFT) exhibited remarkably different execution times during this phase of the investigation. For one, Kalign for MSA yielded regular timeframes of t > 236 s and t < 239 s on eight separate instances, whereas ClustalW and MAFFT required significantly longer timeframes per same number of intervals (t > 459 s and t < 4012 s). Both ClustalW and MAFFT also far exceeded a computationally efficient mark for physical memory usage set by Kalign. These implications are not far reaching, and will be further discussed in the section below. On another quick note, benchmark measurements were executed utilizing a simple script that captures runtime operations in real-time. See additional notes for more details in Bi, S., et al. 2014[15].
To sum up, Kalign is renowned for producing optimal execution times and the following MSA procedure would require minimal computational resources given the selection of sequence type (mtDNA). First, UGENE’s multiple sequence alignment tool Bohle, H.M., et al. 2012[11]. was initiated by importing, processing and combining the genomic datasets [in FASTA format]; one file containing two hundred thirty-five nucleotide sequences, and others containing the remaining family types appropriated for this study. MSA gap penalty scores were modified slightly during successive intervals until an optimal global alignment was achieved. The final set of intervals resulted in a 17,240 base-pair alignment.
Building a phylogenetic tree based on mtDNA sequences
In terms of any tree-building exercise, overall accuracy is directly contingent on the precision of a reliable base-pair alignment output, as highlighted in the section above. Before proceeding, I should mention another important detail relevant to my selection of iterative method for MSA. In a 2005 study, Lassmann & Sonnhammer 2005 noted the following regarding the effectiveness of Kalign for analyzing large-scale base-pair alignments: “The number of input sequences has a big effect on the running time of each method as the complexity of all alignment algorithms depend on it. Conversely, the more sequences that are used in an alignment, the better an alignment algorithm should perform. To our surprise, the quality of all methods except for Kalign decreased when the number of input sequences was increased. The difference in alignment quality between Kalign and the next best method reaches 15% at 400 sequences Lassmann & Sonnhammer, 2005[14].”
Because of its relative speed and the subjective quality of alignments described by Lassmann & Sonnhammer 2005, Kalign generally wins in difficult cases of high evolutionary distance. However, despite Kalign’s superiority in efficiency and resolution for large-scale processing, ClustalW and MAFFT yielded nearly identical outputs with only minor variations. As a result, the computational drawbacks referenced above would not necessarily influence the final renderings nor does it contain statistically significant disagreements with Kalign that should be emphasized further. This may very well be attributed to sequence selection (mtDNA), where light-weight datasets improved accuracy by means of runtime execution. In any such case, minor variations may still have considerable implications in terms of individual placement on phylogenetic trees. For this reason, and others highlighted by Lassmann & Sonnhammer 2005, confidence levels are increased in the base-pair alignments generated by Kalign for MSA.
Where Kalign is proven reliable, PHYLIP neighbor-joining is equally effective in producing highly probable diagrams amid scenarios involving low degrees of variance, regardless of alignment size. Exclusively selected for this tree-building exercise, PHYLIP neighbor-joining is an accurate and statically consistent polynomial-time algorithm that does not assume that all lineages evolve at the same rate, and it constructs a tree by successive clustering of lineages, setting branch lengths as the lineages join [where a set of n taxa requires n – 3 iterations; each step is repeated by (n – 1) x (n – 1)] (Felsenstein, 1981[16] & Ragan, 1993[17]). This method [via UGENE] utilizes a set of default parameters for distance matrix model F84. Additional bootstrapping compilers were not required for this operation, and transition ratios are generated automatically under default settings from Bohle, H.M., et al. 2012[11]. For reference purposes, the following formula demonstrates a standard neighbor-joining Q-matrix algorithm:
[Q (i, j) = (n – 2) d (i, j) – Σ {n, k = 1} d (i, k) – Σ {n, k = 1} d(j, k)] (1)
Pair to node (distances):
[(f, u) = ½ d (f, g) + ½(n – 2) [Σ {n, k = 1} d (f, k) – Σ {n, k = 1} d (g, k)] (2)
Taxa to node (distances):
[d(u,k) = ½ [ d(f,k) + d(g,k) – d(f,g) ] ; (3)
Results
On a broad spectrum, the phylogenetic diagram(s) generated by the PHYLIP neighbor-joining method [coupled with Kalign for MSA] depicts an evolutionary scenario consistent with hierarchal models of mammal class diversification; and it highlights the divergent events that further distinguish the three modern groupings within the class mammalia: monotremes, marsupials and placentals. Sister taxa among clade distributions are arranged in accordance with morphological classifications. Each taxonomic unit with descendants also constitutes the inferred most recent common ancestor of the descendants and the edge lengths may be interpreted as rough time estimates.
Located on the internal branches near the root, we find nodes that represent the oldest common ancestor of Cretaceous mammals, presumably. As we move from one internal node to the next, the variation of genetic distance increases minutely and only widens at the genera level. Similarity ratios show a marginal disparity – ranging between + - 65 to + - 92 percent – among seven family types concentrated nearest the root; and it shows a + - 50 to + - 64 percent divergence from taxonomic units within the remaining clades. The identity distance matrices shown on Figure 1 & Figure 2 better illustrate the measurements of genetic divergence between each sequence, where the final distance value is the average of PHYLIP neighbor-joining estimates. In the context of extinction events and the ecological niches that are filled thereafter, we might expect to observe widening gaps in degrees of genetic variation following a period of rapid diversification. Here, this occurs on several different scales.
Maximal node measurements further support that interpretation. Estimates of divergence are not always straightforward and the rate of evolution is not uniform in different lineages Ramnauth, 2013[18]. Nonetheless, it is widely held that trees exerting node measurements of 0.75 or higher are generally reliable, in terms of Bayesian-obtained approximates Douady et al., 2003[19]. A high value means that there is strong evidence that the sequences to the right of the node cluster together to the exclusion of any other Ramnauth, 2013[18]. Arguably so, measurements that do not meet a minimal requirement for posterior probabilities in maximal node measurements can lead to erroneous inferences Huelsenbeck et al., 2004[20] & Hall et al., 2007[21]. In this case, the maximal measurement values show a steady rate of calibration consistency, represented by the maximal support measurement per internal node illustrated on (Figure 3). Each maximal support measurement is 1.0, regardless of node placement within the tree. As such, the resulting cladogram shows localized diversification events in multiple time periods, across multiple scales.
Exact time estimates, however, cannot be determined by these figures alone. The molecular clock, shown here by the internal branch lengths and their respective node ages, suggests that placental lineages are far older than traditional models presume; and their diversification may be linked prior to the breakup of the continents before the end of the Cretaceous period Luo et al., 2011[4]. At any rate, the genomic data needed to narrow the scope further was not readily available to this investigation.
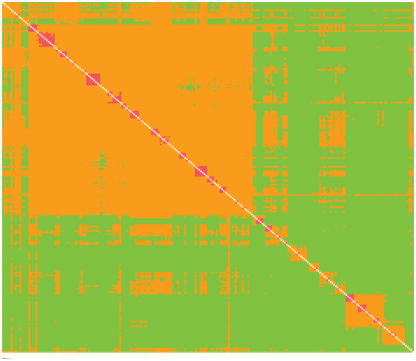
Figure 1: Identity distance matrix (235 sequences; 62 families; 17,240bp)

Figure 2: Identity distance matrix (17 sequences; 7 families; 18,610 bp)
Legend::
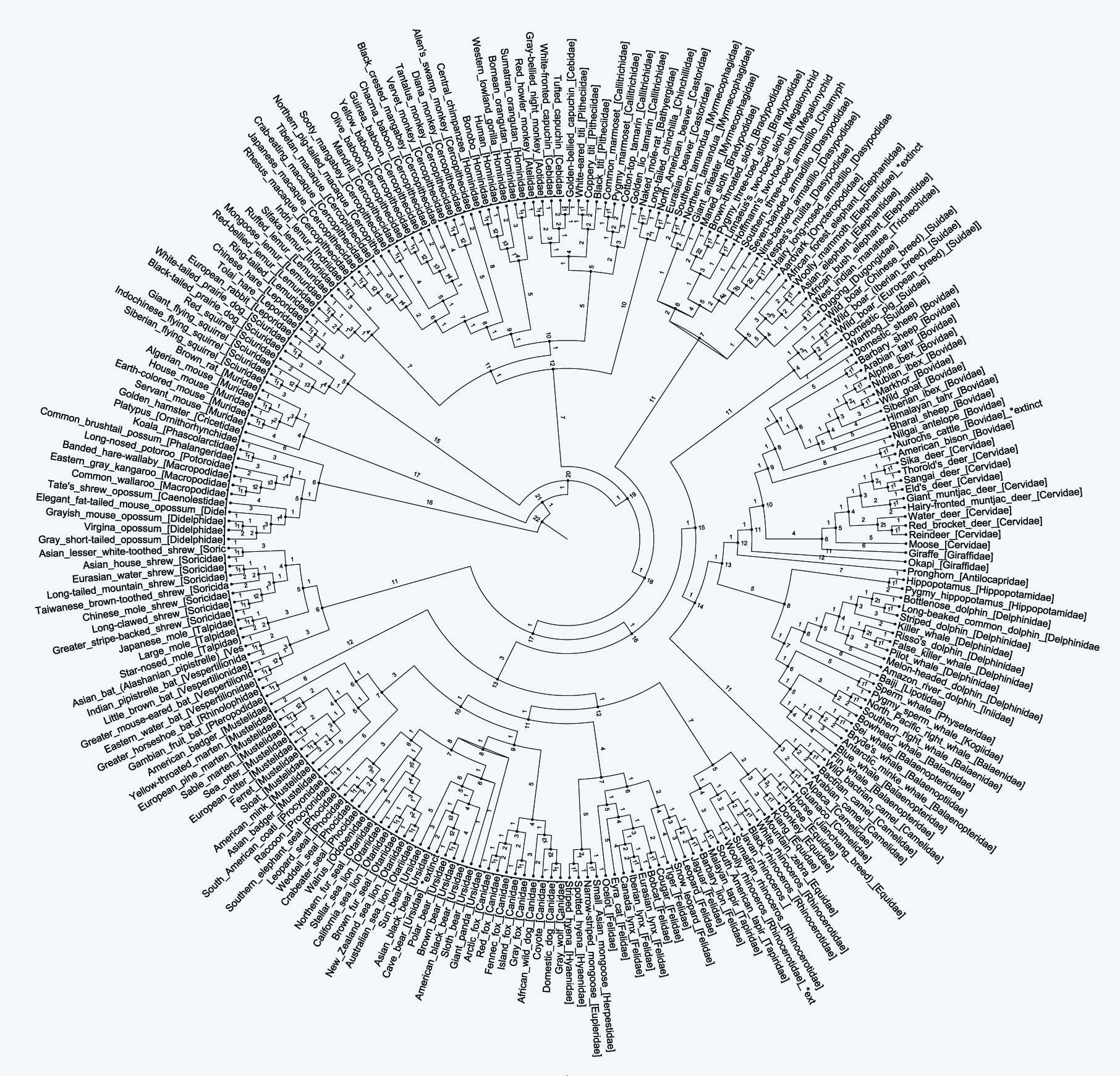
Figure 3: Complete Phylogenetic Tree of Sixty-Two Distinct Mammalian Families Based on Complete mtDNA Sequences (17,240 bp). Includes [in alphabetical order]: Aotidae, Atelidae, Balaenidae, Balaenopteridae, Bathyergidae, Bradypodidae, Bovidae, Caenolestidae, Callitrichidae, Camelidae, Canidae, Castoridae, Cebidae, Cercopithecidae, Cervidae, Chinchillidae, Chlamyphoridae, Cricetidae, Dasypodidae, Delphinidae, Didelphidae, Dugongidae, Elephantidae, Equidae, Eupleridae, Felidae, Giraffidae, Herpestidae, Hippopotamidae, Hominidae, Hyaenidae, Indriidae, Iniidae, Kogiidae, Lemuridae, Leporidae, Lipotidae, Macropodidae, Megalonychidae, Muridae, Mustelidae, Myrmecophagidae, Ornithorhynchidae, Orycteropodidae, Otariidae, Phalangeridae, Phascolarctidae, Phocidae, Pitheciidae, Procyonidae, Physeteridae, Pteropodidae, Rhinocerotidae, Rhinolophidae, Sciuridae, Soricidae, Suidae, Talpidae, Tapiridae, Trichechidae, Ursidae, Vespertilionidae.
Discussion
The “explosive model” of mammal evolution proposes that late placental lineages emerged and diversified to fill niches left vacant after the KT catastrophe O’Leary et al., 2013[22]. Conversely, the cladogram illustrated above (Figure 3) depicts a rather different trajectory. My results suggest that placental lineages are possibly older than traditionally presumed, hinting their diversification was linked prior to the breakup of the continents before the end of the Cretaceous period Luo et al., 2011[4]. Although that interpretation is not new to the field of phylogenetics, it is an independent confirmation of the inferences raised by others; most notably Bi et al. 2014[15] & Luo et al. 2011[4], which reinforce the early divergence of mammals.
As shown on (Figure 3), seventeen distinct organisms belonging to four family types represent the closest living lineages from which placental mammals and marsupials diverge from a common ancestor; including Cricetidae, Muridae, Soricidae and Talpidae. This group of organisms is interesting because of its retention of primitive traits, similar to Juramaia sinensis; the oldest eutherian fossil species ever found Luo et al., 2011[4] & O’Leary et al., 2013[22]. Lines of anatomical evidence support the idea that Juramaia sinensis is closely related to placental mammals O’Leary et al., 2013[22]. For example, Juramaia sinensis has three molars and five pre-molars — like placentals, but unlike marsupials which have four molars and three pre-molars Luo et al., 2011[4]. Before the discovery of Juramaia sinensis, the earliest known fossil relatives of placentals dated to around 125 Myr O’Leary et al., 2013[22]. Since each lineage clearly existed as a distinct entity approximately 125 Myr, the divergence between placentals and marsupials may have occurred sometime before then, presumably Luo et al., 2011. [4]
My findings are not necessarily indicative of a direct living lineage to a common ancestor of a placental in the form a single family type, but could also be explained by missing ancestral unit(s) not present on the cladogram. Other phylogenies, most notably those reconstructed on morphology, often disagree with respect to time estimates and divergence in mammal evolution. But unlike other models that suggest explosive evolution in post-Cretaceous context, the scenario outlined by my results predict a series of rapid diversification events in multiple time periods, and should therefore not be confused with a single “explosive” radiation event preceding or following the K–Pg boundary.
Lastly, something should be said about the sequence selection in phylogeny research. A handful of studies have well documented the limitations of using mtDNA to reconstruct phylogenies that involve time-extended lineages. Rapid mutation rates in mtDNA produce significant molecular variance among immediate populations Excoffier et al., 1992. This has notable benefits when studying ancestral relationships whose divergence times are thought to be no greater than 8 to 10 Myr DeSalle & Giddings, 1986[24]. However, in reciprocated cases, efficacy may become less resolved beyond that scope. In addition to that, hybridization effects can cause mtDNA to move freely between species, which could infer incorrect relationships when building phylogenies Hurst & Jiggins, 2005[25].
Despite these legitimate critiques, this paper addressed the practical applications of using mitochondrial datasets in large-scale studies. I would also point out that examples of natural hybridization leading to speciation are exceedingly rare, especially in mammals Arnold et al., 1997[13]. While most known cases of hybrid speciation occur in plants, the majority of documented cases involving animals have been observed in fish and insects Larsen et al., 2010[26]. Perhaps most important in this discussion, mtDNA datasets have repeatedly shown to provide enough sufficient resolution for reconstructing a robust phylogeny and it also facilitates the molecular dating of divergence events within a phylogeny Krause et al., 2008[27]. Mitochondrial DNA is particularly useful in phylogenetic studies, as it demonstrates high interspecies conservation and at the same time is variable enough to allow intraspecies differentiation Rutty, 2016[28].
Conclusion
By assessing the number of node measurements, where the taxonomic units with higher nucleotide substitution rates reside on the far ends of internal branches, and by interpreting the molecular clock in accordance with their respective node ages, I find very good support for my original inquiry. Based on this interpretation, the resulting cladogram provides evidence for localized diversification events in multiple time periods, across multiple scales. This inference also qualifies the second part of this study, which holds that the closest living lineages of early placentals derive from a specific group that shares homologous traits with the oldest eutherian fossil species ever found. The lack of data needed to narrow the scope further makes an accurate time estimate difficult to acquire; but these results may provide a reliable starting point for a more complete systematic investigation.
Additional Notes
1Annotations: NC_021398.1, KJ920198.1, KF696672.1, KJ131179.1, KJ545899.1, AF348081.1, AB061527.1, KU246040.1, KM503097.1, KM092492.1, KU144678.1, KT934322.1, AB099482.1, EU333163.1, EU117375.1, FJ236998.1, KF938324.1, KF938321.1, KF938330.1, KF938327.1, NC_009510.1, FJ207526.1, FJ207523.1, FJ207525.1, FJ207527.1, JX101652.1, KT290893.1, FJ207531.1, FJ207529.1, HQ832482.1, NC_018595.1, NC_014701.1, NC_006993.1, HM138200.1, JN399997.1, HM049636.1, FJ705435.1, AY239042.1, JF802125.1, JN632595.1, KP405229.1, JN632657.1, KM506758.1, KT998647.1, NC_020476.1, NC_001788.1, NC_012682.1, NC_001808.1, NC_001779.1, NC_012683.1, NC_012684.1, KJ417810.1, AJ428947.1, JN601075.1, AP003428.1, AF492350.1, NC_013996.1, GU947006.1, AP003425.1, JN632625.1, KF776494.1, EF551003.1, EF551002.1, KM236783.1, KJ866876.1, NC_016470.1, KP202279.1, NC_027083.1, NC_028319.1, NC_028313.1, AY873843.1, NC_020670.1, JF894376.1, KJ419916.1, KP202284.1, KP202282.1, KP202258.1, KF857179.1, AY729880.1, NC_008093.1, NC_028427.1, NC_026723.1, KP129082.1, KJ603240.1, JN711443.1, KP342451.1, NC_009126.1, KU052604.1, KU146454.1, HM106331.1, HM106330.1, KM091450.1, KJ202625.1, KC660129.1, EF672696.1, AB291077.1, KM347744.1, NC_003426.1, HQ685964.1, AJ428577.1, NC_009971.1, FM177765.1, EU327344.1, EF196662.1, NC_004023.1, AM181021.1, AM181020.1, AM181019.1, AM181018.1, AM181017.1, GU475464.1, NC_004029.2, AM181023.1, AM181024.1, AM181025.1, AM181016.1, AM181026.1, AY075116.1, AM904728.1, FJ905814.1, X97337.1, GU734783.1, NC_000934.1, KJ557424.1, AP008987.1, DQ316068.1, EU155210.1, NC_001808.1, NC_002078.1, FJ905813.1, KP789021.1, KJ417810.1, JX312732.1, NC_028567.1, NC_009629.2, KU168760.1, JN632597.1, DQ409327.1, EF536350.1, JN632608.1, AJ566364.1, EU681954.1, KT818545.1, KT818547.1, Y11832.1, KT818546.1, KT818553.1, NC_028572.1, KT818551.1, KT818552.1, KT818538.1, KT818537.1, KT818523.1, KR336791.1, KT818524.1, NC_006299.1, NC_001610.1, NC_029381.1, NC_005825.1, AJ508400.1, NC_000891.1, NC_001794.1, AF357238.1, AJ639873.1, NC_008133.1, AJ421451.1, NC_026098.1, NC_026085.1, AM905040.1, KJ944188.1, KJ944181.1, NC_005943.1, KM401548.1, KM360179.1, KM851031.1, KP330231.1, EU294187.1, KT159932.1, KC757403.1, NC_020006.2, NC_020010.2, NC_020009.2, HM068590.1, NC_001644.1, NC_011120.1, NC_001646.1, HM156696.1, KU353725.1, NC_009747.1, KC757391.1, KJ434958.1, KJ434962.1, JX946199.2, KC757401.1, EF597502.1, NC_027973.1, NC_029346.1, NC_029191.1, NC_029849.1, NC_029375.1, NC_020326.1, NC_029939.1, NC_015112.1, NC_019612.1, NC_026443.1, JX572159.1, JQ743657.1, KF570389.1, EU557094.1, EU557097.1, KF570335.1, JF289177.1, EU557095.1, JF289176.1, JF289173.1, NC_023889.1, NC_019578.2, NC_007629.1, NC_005276.1, JN632624.1, KC312610.2, AJ554055.1, AP006474.1, AJ554051.1, AP006473.1, AP006470.1, AB201258.1, AP006466.1, KC572829.1, NC_001601.1, FR691684.1, FR691688.1, NC_013276.1, NC_026706.1, NC_021386.1, NC_010638.1, KX084803.1, NC_009492.1, NC_002369.1, KM114606.1, EU450583.1, NC_010650.1, NC_025952.1, NC_026705.1
2Benchmarking for Multiple Sequence Alignment (code summarized): :: Filename: timecmd.bat
:: Author: Tommy Rodriguez
@echo off
@setlocal set start=% time %
:: runs command for ugeneui.exe (Kalign, MAFFT & ClustalW)
ugene align-kalign – in = FILE PATH – out = FILE PATH set end = % time % set options = “tokens =1-4 delims=:.”
cmd /c ugene align-mafft –in = FILE PATH – out = FILE PATH set end = % time % set options = “tokens=1-4 delims=:.”
cmd /c ugene align --in=FILE PATH --out=FILE PATH set end = % time % set options =”tokens =1- 4 delims=:.”
for /f % options% %% a in (“%start%”) do set start_h = %% a & set /a start_m =100%% b %% 100 & set /a start_s=100 %% c %% 100 & set /a start_ms=100%% d %% 100 for /f % options% %% a in (“%end%”) do set end_h = %% a&set /a end_m =100 %% b %% 100 & set /a end_s =100 %% c %% 100 & set /a end_ms=100%%d %% 100 set /a hours = %end_h%-%start_h% set /a mins = %end_m% -% start_m% set /a secs=%end_s%- %start_s% set /a ms = %end_ms%-%start_ms% if %hours% lss 0 set /a hours = 24%hours% if %mins% lss 0 set /a hours = %hours% - 1 & set /a mins = 60% mins% if %secs% lss 0 set /a mins = %mins% - 1 & set /a secs = 60%secs% if %ms% lss 0 set /a secs = %secs% - 1 & set /a ms = 100%ms% if 1%ms% lss 100 set ms = 0%ms%
echo [method] took %hours%:%mins%:%secs%.%ms% (%totalsecs%.% ms%s total)
3UGENE was used in comparative sequence analysis. The DNA sequences noted above are in FASTA format. They were obtained from the NCBI database archives.
References
- 1. Vié, J.C., Hilton-Taylor, C., Stuart, S.N. Wildlife in a changing world: an analysis of the 2008 IUCN Red List of threatened species. (2009) IUCN.
- 2. Vaughan, T.A., Ryan, J.M., Czaplewski, N. J. Mammalogy. (2013) Jones & Bartlett Publishers.
- 3. Robertson, D.S., McKenna, M.C., Toon, O.B., et al. Survival in the first hours of the Cenozoic. (2004) GSA Bulletin 116(5-6)
- 4. Luo, Z.X., Yuan, C.X., Meng, Q.J., et al. A Jurassic eutherian mammal and divergence of marsupials and placentals. (2011) Nature 476(7361): 442-445.
- 5. Meredith, R.W., JaneÄ










