
Isolated Mitochondrial Myopathy Misdiagnosed as Polymyositis
Yong Seon Choi1, Ji Yeong Chae1, Hyung Woo Kim1, Jae Min Lee1, Jung Ho Won1, Sun Hong You1, Hyun-Jeung Yu2, Eun Hye Jeong2*
Affiliation
- 1Department of Internal Medicine, Bundang Jesaeng General Hospital, Seongnam, Korea
- 2Department of Neurology, Bundang Jesaeng General Hospital, Seongnam, Korea
Corresponding Author
Eun Hye Jeong, M.D., Department of Neurology, Bundang Jesaeng General Hospital, 20, Seohyeon- ro180beon-gil, Bundang-gu, Seongnam-si, Gyeonggi-do, Korea, E-mail: gracej3@naver.com
Citation
Jeong, E.H. et al. Isolated Mitochondrial Myopathy Misdiagnosed as Polymyositis. (2016) Int J Neuro Brain Dis 3(4): 1- 4.
Copy rights
© 2016 Jeong, E.H. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Mitochondrial myopathy; Polymyositis
Abstract
Mitochondrial myopathies are caused by dysfunction of the mitochondria and are systemic disorders that present with diverse clinical manifestations. They have been found to be especially prevalent in children. In this report, we present the case of a 55-year-old male patient who developed progressive symmetric proximal muscle weakness in both upper and lower extremities. Although the elevated levels of muscle enzymes and the findings of magnetic resonance imaging of lower extremities were suggestive of polymyositis, we made a final diagnosis of isolated mitochondrial myopathy based on the result of the biopsy of the thigh muscle. In this report, we emphasize the importance of performing a correct differential diagnosis of myopathies, particularly in the cases without evidence of involvement of other organs.
Introduction
Mitochondria are intracellular organelles that host the components of the respiratory chain and produce adenosine triphosphate. They play an integral part of the metabolism in the body, and contain maternally-inherited mitochondrial DNA (mtDNA). Mutations in these genes and in nuclear DNA impair the function of nerves, muscles, the brain, the liver, and the endocrine system, which are heavily reliant on aerobic metabolism. Mutations in mtDNA and nuclear DNA lead to mitochondrial disorders with heterogeneous and polymorphic pathological manifestations[1]. The clinical expression of mitochondrial myopathy in the affected skeletal muscle may vary in terms of severity from mild exercise intolerance to fatal infantile encephalomyopathy or multisystemic dysfunction.
Mitochondrial myopathies include progressive external ophthalmoplegia; Kearns-Sayre syndrome; mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS); myoclonus, epilepsy, and ragged red fibers (MERRF); ataxia-neuropathy syndromes, and isolated myopathy. Since mitochondrial myopathies comprise such a heterogeneous group of disorders, each with its own distinctive symptoms and clinical characters, diagnosing these disorders accurately and promptly is difficult[2]. Moreover, in case of isolated myopathy, differential diagnosis with other causes of myopathies is sometimes challenging because of absence of other systemic manifestations. We report a case of isolated mitochondrial myopathy confirmed by muscle biopsy, initially misdiagnosed as polymyositis.
Case Report
55-year-old male patient was admitted to the hospital with progressive general weakness and fatigue 3 months before. The patient had a medical history of pulmonary tuberculosis 25 years ago and had remained remission-free ever since. There were no other notable medical histories including seizure or stroke, and the patient was not on any medication. He was a florist and reported neither a personal history of smoking or drinking nor a family history of neuromuscular disorders or hereditary diseases. The patient complained of general weakness and early fatigue while walking, especially with difficulty in ascending stairs, rising from supine or sitting position, and raising his arms above his head. Physical examination showed a blood pressure of 98/65 mmHg, a heart rate of 92 beats per minute, a respiration rate of 18 breaths per minute, and a body temperature of 36.5°C. His height was 167 cm, and weight was 56.7 kg. A clinical assessment of the head, neck, chest, abdomen, and skin did not reveal any particular signs. Neurological examination demonstrated that the cranial nerve functions were normal with no ptosis or ophthalmoparesis, but the muscle strength of both shoulders and hips was weakened to Medical Research Council grade IV. Although his toe and heel gait motions were normal, squatting was impaired. No sensory abnormalities were noted, and also there were no abnormalities in the deep tendon reflex and cerebellar function.
Peripheral blood tests showed that white blood cell count was 11.8 × 103/L, the hemoglobin level was 10.4 g/dL, and the platelet count was 508 × 103/L. The erythrocyte sedimentation rate was 120 mm/hr, the blood urea nitrogen level was 14 mg/dL, and the creatinine level was 0.76 mg/dL. Liver function tests found a level of aspartate transaminase of 267 IU/L, an alanine aminotransferase level of 143 IU/L, a total bilirubin level of 0.40 mg/dL, an alkaline phosphatase level of 256 IU/L, and a gamma glutamic transpeptidase level of 23 IU/L. Viral hepatitis markers were within the normal range. The level of creatine phosphokinase was elevated to 6,531 IU/L and the level of creatine kinase isoenzyme elevated to 67.49 ng/mL, but otherwise the results of blood coagulation tests and urinalysis were normal. Arterial blood gas analysis under subatmospheric pressure results were: pH, 7.456; pO2, 73.8 mmHg; pCO2, 39.6 mmHg; and HCO3-, 27.3 mmol/L. When screened for autoimmune antibodies, the patient was positive for anti-nuclear antibodies and anti-SSA antibodies. Negative results were found for anti-ribonucleo protein antibodies, anti-Smith antibodies, anti-Jo-1 antibodies, anti-Scl 70 antibodies, and anti-neutrophil cytoplasmic antibodies. The result of electrocardiography was normal and plain chest radiography did not show any abnormal signs.
The patient showed progressive symmetric proximal muscle weakness in both extremities, so we suspected myopathy and performed nerve conduction study and electromyography (EMG). The result of nerve conduction study was normal and the EMG on the right upper and lower extremities revealed denervation potentials in the biceps brachii, triceps brachii, flexor carpi ulnaris, vastus lateralis, tibialis anterior and paralumbosacral (L4-5, S1) muscles, suggestive of active-stage myopathy. The results of magnetic resonance imaging (MRI) of lower extremities showed diffuse symmetric signal changes and mild edema with enhancement of bilateral thighs and pelvic muscles [Figure 1]. Depending on the clinical presentation of progressive symmetric proximal muscle weakness, laboratory findings including elevated muscle enzymes and erythrocyte sedimentation rate, the results of EMG and MRI, we initially diagnosed as polymyositis. For confirmation of the diagnosis, we performed muscle biopsy of the right rectus femoris muscle. Due to the time period before biopsy results, the patient received corticosteroid and showed some improvement of the symptoms. However, the result of the biopsy showed ragged red fibers in modified Gomori trichrome stain and abnormal mitochondria with parking lot inclusions on electron microscopy (EM), suggestive of mitochondrial myopathy [Figure 2]. In order to assess the possibility of other organ involvement and to evaluate possible complications, we performed a biopsy of the liver, but found normal findings. Likewise, ophthalmologic examination, echocardiogram, electroencephalography (EEG) and brain MRI all returned normal findings. In order to evaluate the possibility of other mitochondrial disorders, we tested the patient’s blood for the MT-TL1 and MT-TK genes, which are known to be related with MELAS and MERRF, and found that the results were negative. Therefore, we finally diagnosed as isolated mitochondrial myopathy, and administered coenzyme Q10 and carnitine to the patient.
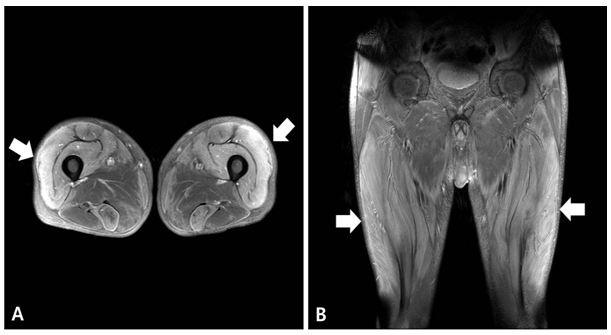
Figure 1: Gadolinium-enhanced T1-weighted axial (A) and coronal (B) images of magnetic resonance imaging of lower extremities reveal diffuse symmetric signal changes and mild edema with enhancement in muscles of bilateral thighs (quadriceps femoris group, semitendinosus, sartorius, gracillis muscles) and pelvis (obturator internus and externus muscles) (arrows).

Figure 2: Electron microscopy of muscle biopsy shows swollen mitochondria with parking lot inclusions(arrows).
Discussion
A notable feature of muscular disorders is that they present with common clinical presentations, symptoms, and laboratory findings, which makes it important to differentiate mitochondrial myopathy, inflammatory diseases, and degenerative diseases when diagnosing and treating muscular disorders. With myopathy, physical and laboratory investigations by themselves cannot be used to diagnose the condition conclusively. A muscle biopsy must be carried out in order to obtain a definitive diagnosis. Although the primary dysfunction in mitochondrial myopathy is at the level of the mitochondria, it presents as a systemic disease with clinical symptoms involving a broad range of systems, usually affecting the nervous system and muscles, both of which include components that consume a large amount of energy.
The causes of mitochondrial myopathies include mutations in mtDNA, defects in nuclear DNA sequences that encode proteins that are translated in the mitochondria, dysregulated interaction between mtDNA and nuclear DNA, and the direct effects of drugs such as zidovudine and clevudine. Mitochondrial disorders can be classified by genotype. The heterogeneity of genetic defects leads to variation in clinical symptoms among individuals. The genetic defects of mitochondrial myopathies can be classified according to the type of DNA mutation: deletions in mtDNA, duplication of mtDNA, point mutations in mtDNA, or other mutations in nuclear DNA. Mutations in genes relating to the replication of mtDNA or to respiratory chain reactions, such as the DNA polymerase gamma catalytic subunit and mtDNA genes encoding cytochrome b (MTCYB), thymidine kinase 2 (TK2), and cytochrome oxidase (COX), have been implicated in mitochondrial myopathy[3,4].
Mitochondrial myopathies are clinically and genetically diverse. This diversity is attributed to the range of mitochondria that can elicit the symptoms of these disorders. Within mitochondrial mutations, duplication and point mutations of mtDNA are inheritable, but deletion mutations are not. This is the reason for which a high level of genetic heterogeneity, which refers to the mixture of mutated and non-mutated genes, is seen among individuals[5]. Since hundreds to thousands of mitochondria exist in a cell and, within a single mitochondrion, only a subset of the several mtDNA that exist experience mutations, upon the somatic division of cells, the degree of mutation in the mtDNA differs substantially among cells: This concept is described as heteroplasmy.
Therefore, the differences in the mutational load of the mtDNA at the cellular level that occur as a result of heteroplasmy account for the differences in the severity of the symptoms among individuals. In general, it has been shown that the symptoms of mitochondrial myopathy manifest when the proportion of mutated mtDNA is over 85%. Interestingly, it has been shown that the effect of the same mutational load may differ among organs and tissues within a patient. Not only can the same mutational load lead to differences in the symptoms that are experienced, it can also lead to differences among patients that explain the variability in clinical presentation. Studies have attempted to identify a relationship between the heteroplasmy of mtDNA in the blood and the severity of symptoms, but no conclusive correlation has yet been found[6].
According to a previous case report, a case of mitochondrial myopathy mimicking polymyositis with increased level of muscle enzymes was finally diagnosed with Kearns-Sayre syndrome confirmed by muscle biopsy and genetic analysis[7]. In our patient, the results of the study including the elevated level of muscle enzymes and the erythrocyte sedimentation rate, positive antinuclear antibodies, and the MRI suggestive of myositis were indicative of inflammatory myopathy such as polymyositis. However, the result of muscle biopsy performed to confirm our diagnosis showed ragged red fibers in modified Gomori trichrome stain and swollen mitochondria with parking lot inclusions on EM, suggestive of mitochondrial myopathy.
In general, a conclusive diagnosis of mitochondrial myopathy can be made if a biopsy shows ragged red fibers in Gomori trichrome stain or if EM shows an abnormal proliferation of mitochondria, enlarged mitochondria, or the presence of parking-lot inclusion bodies. Furthermore, when muscle biopsy results are inconclusive for diagnosis, biochemical approaches can help to detect defects in the components of the mitochondrial respiratory chain, such as complex I deficiency, complex II deficiency, or complex IV deficiency, and molecular genetic approaches such as sequencing of the genome to detect genetic defects or mutations can make a valuable contribution[1,2]. In the present case, unfortunately we could not corroborate our diagnosis through additional biochemical or genetic tests providing a more detailed assessment of the pathogenesis of the condition, but we thought that the histological findings with special staining and EM were sufficient to drive the correct diagnosis. In general, mitochondrial myopathy patients present with clinical features of multi-organ dysfunction, and rarely present with isolated symptoms. In our case, bilateral symmetric proximal muscle weakness of the extremities was the only symptom. We excluded the possibility of coexisting dysfunction of other organs by several special studies such as EEG, brain MRI, cardiac evaluation, and liver biopsy, so we could make a final diagnosis of isolated mitochondrial myopathy.
Unfortunately, no available treatments target the fundamental cause of mitochondrial myopathy, and no consensus exists for the gold standard of treatment. Studies have shown that the supplementation of respiratory chain components such as coenzyme Q10 and L-carnitine is beneficial, which has led to their use in treatment. In recent years, since dichloroacetate has been shown to reduce toxic metabolites, such as lactic acid, it has been used as an experimental agent. Similarly, antioxidants such as vitamin K3, vitamin C, riboflavin, and thiamine have been tested, but yet have not shown significant clinical value[8,9]. In addition, several international and national level reports have shown that patients with mitochondrial disorders, specifically MELAS, have responded to corticosteroid treatment, as described in our case. However, the exact mechanism by which steroids act on patients with mitochondrial disorders is unknown. According to some reports, steroids have been shown not only to exert their effect in the nucleus but also in the mitochondria, because glucocorticoid receptors have been found in the mitochondria. This means that steroids may exert their effect on the pathogenesis of disease by acting on the receptor, on the nuclear DNA, or even cooperatively. Furthermore, steroids have been shown to directly stimulate mitochondrial transcription, thereby exerting a beneficial effect by influencing the biosynthesis of the respiratory chain[10,11].
Mitochondrial myopathies are commonly associated with other neurological symptoms, and in most patients, other organs are already affected by the time of diagnosis. However, for cases of isolated myopathy such as described in our case, since the isolation of the condition within a single organ must be verified through a process of elimination, a more discriminatory approach is required, which explains the need of performing a muscle biopsy.
In this case report, the authors initially misdiagnosed a patient’s illness at the time of admission as polymyositis, however, finally diagnosed as isolated mitochondrial myopathy after muscle biopsy. Thus, a postulated diagnosis of myopathy must be confirmed through a biopsy of the tissue of concern.
References
- 1. Milone, M., Wong, L. J. Diagnosis of mitochondrial myopathies. (2013) Mol Genet Metab 110(1-2): 34-41.
- 2. Pfeffer, G., Chinnery, P.F. Diagnosis and treatment of mitochondrial myopathies. (2013) Ann Med 45(1): 4-16.
- 3. DiMauro, S. Mitochondrial myopathies. (2006) Curr Opin Rheumatol 18(6): 636-641.
- 4. Shin, J.H., Kim, D.S. Genetics of mitochondrial myopathies. (2013) J Genet Med 10(1): 20-26.
- 5. Chinnery, P., N., Andrews, R.M., et al. Clinical mitochondrial genetics. (1999) J Med Genet 36(6): 425- F., Howell 436.
- 6. Mehrazin, M., Shanske, S., Kaufmann, P., et al. Longitudinal changes of mtDNA A3243G mutation load and level of functioning in MELAS. (2009) Am J Med Genet A 149A(4): 584-587.
- 7. Corrado, A., Cantatore, F.P., Serlenga, L., et al. Mitochondrial disease mimicking polymyosities: a case report. (2002) Clin Rheumatol 21(5): 411-414.
- 8. DiMauro, S. Mitochondrial encephalomyopathies: what next? (1996) J Inherit Metab Dis 19(4): 489-503.
- 9. De Stefano, N., Matthews P. M., Ford B., et al. Short-term dichloroacetate treatment improves indices of cerebral metabolism in patients with mitochondrial disorders. (1995) Neurology 45(6): 1193-1198.
- 10. Finsterer J., Frank M. Glucocorticoids for mitochondrial disorders. (2015) Singapore Med J 56(2): 122-123.
- 11. Hedley-Whyte, E.T., Hsu, D.W. Effect of dexamethasone on blood-brain barrier in the normal mouse. (1986) Ann Neurol 19(4): 373-377.










