
MAD2 in the Spotlight as a Cancer Therapy Regulator
Bargiela-Iparraguirre, J.1,2, Fernandez-Fuente, M, Calés C1, Herrera, L.A2 and Sanchez- Perez, I1,2*
Affiliation
- 1Departament de Bioquímica, Facultad Medicina, Instituto de Investigaciones Biomédicas Madrid CSIC-UAM; Madrid, Spain
- 2Unidad de Investigación Biomédica en Cáncer, Instituto Nacional de Cancerología (INCan)-Instituto de Investigaciones Biomédicas (IIB), Universidad Nacional Autónoma de México (UNAM), México; Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Circuito Escolar S/N, Ciudad Universitaria, Coyoacán, México
Corresponding Author
Isabel Sanchez-Perez, PhD, Departament de Bioquímica, Facultad Medicina, Instituto de Investigaciones Biomédicas Madrid CSIC-UAM; C/Arturo Duperier 4, 28029 Madrid, Spain, Tel: (+34) 91-5854380/ Fax: (+34) 91- 58544001, E-mail: misanchez@iib.uam.es ; is.perez@uam.es
Citation
Sanchez- Perez, I., et al. MAD2 in the Spotlight as a Cancer Therapy Regulator. (2016) Int J Cancer Oncol 3(2): 1-6.
Copy rights
© 2016 Sanchez- Perez, I. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
MAD2; Chemotherapy; Cell cycle; Mitosis; Cancer; SAC; Biomarker; Resistance
Abstract
MAD2 is a key protein required for mitotic checkpoint function and for the maintenance of accuracy on the mitotic process of every cell cycle. Deregulation of MAD2 is associated with both progression and poor prognosis in cancer disease. However, new evidence highlights the implication of MAD2 in other biological functions besides its classical Mitotic checkpoint implication, such as apoptosis, senescence and also DNA damage repair, which can ultimately define the response to a specific treatment. Development of novel therapeutic approaches and optimization of the existing therapies are now required for the design of successful treatments of cancer. MAD2 is emerging as a new key target for the design of more effective and personalized treatments in cancer disease.
Introduction
Mitosis is the last chance that cells have to avoid chromosome miss-segregation, and this can be achieved thanks to the activation of the spindle assembly checkpoint (SAC). SAC is a conserved mechanism, which ensures the fidelity of chromosome distribution in mitosis by preventing anaphase onset until the correct bipolar microtubule-kinetochore attachments are formed. SAC controls Metaphase-Anaphase transition and delays anaphase until correct bipolar attachment of sister chromatids to the mitotic spindle is achieved, thus ensuring accuracy of the chromosome segregation in mitosis[1]. This complex of proteins includes MAD1, MAD2, BUB1, BUB3, BUBR1, in association with the APC/C activator Cdc20[2]. When SAC is activated, it inhibits the anaphase-promoting complex/cyclosome (APC/C), ligase complex which induces degradation of specific cell cycle regulatory proteins, such as securin, an inhibitor of anaphase initiation activation, and Cyclin B. Degradation of this proteins ultimately leads to mitosis exit[3,4].
MAD2 has a central role in the modulation of the mitotic checkpoint: it activates the “waiting signal” when the microtubules are not correctly attached to the kinetochore in order to prevent unequal chromosome segregation[5]. The gene MAD2L1 (mitotic arrest deficient-like 1) localized in 4q27 encodes MAD2, a small protein that is highly conserved from yeast to humans. The assembly and consequent activation of the Mitotic Checkpoint Complex (MCC) is initiated by the conversion of MAD2 from an open (O-MAD2) to a closed (C-MAD2) conformation, which then binds tightly to Cdc20. The C-MAD2–Cdc20 (MC) subcomplex associates then with BUBR1-BUB3 to form the mitotic checkpoint complex MCC[6]. Conversely, the disassembly of MCC that takes place when the checkpoint is turned off leads to the conversion of C-MAD2 back to O-MAD2[7,8]. Recently, a MAD2-interacting protein, p31comet, has been characterized as a spindle checkpoint silencer during mitosis[9-11]. This protein is a structural mimic of O-MAD2 (open state) and binds to C-MAD2 (closed state). Binding of p31comet prevents dimerization of MAD2, which in turn inhibits the conformational change of O-MAD2 into C-MAD2, its active form, and ultimately triggers the exit from the mitotic process[9,10]. (FIGURE 1)
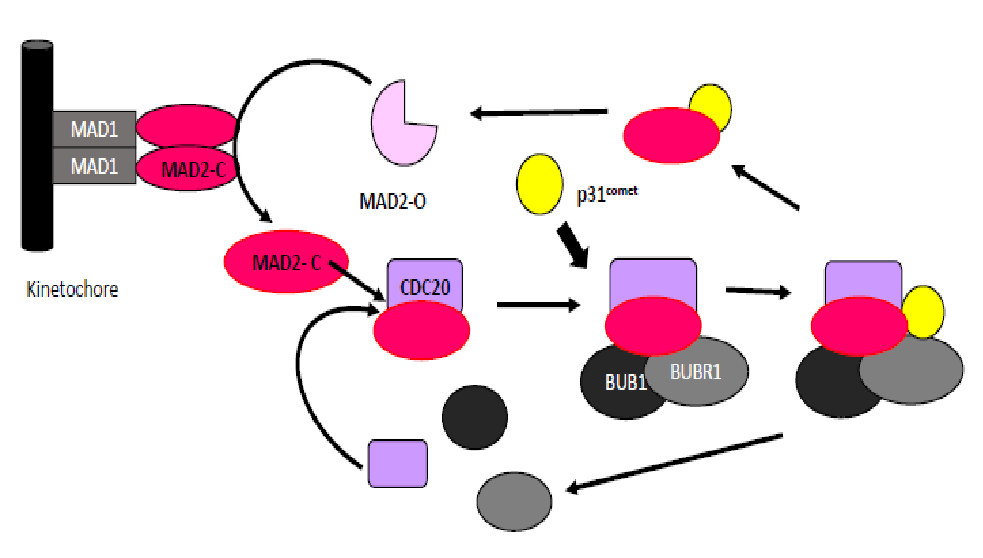
Figure 1:
The aim of this review is to highlight the relationship between MAD2 protein levels and chemotherapy response. Although the role of MAD2 as a prognostic factor in solid tumors is now well established, the data we analyze here suggest that MAD2 could be controlling other pathways apart from controlling SAC. These new functions could drive the ultimate fate of the cell: apoptosis, senescence or DNA repair, as a consequence of the treatment with drugs routinely included in the therapeutic regimen for patients. After careful analysis of this information, we discuss here the role MAD2 as a potential new therapeutic biomarker.
Deregulation of MAD2 levels in cancer and therapy response in vitro and in vivo
Deregulation of MCC protein level increases chromosome instability (CIN), a well known hallmark of cancer. Overexpression MAD2 is commonly found in gastric, colorectal, lung, breast and human ovary tumors, and many authors have demonstrated its correlation with tumorigenesis invasion and metastasis[12,13]. Using rodent models of the disease, it has been shown that reduced levels of MAD2[14], cause a mild increase in the onset of spontaneous tumors (around 30%). On the other hand, after generation of mice over-expressing MAD2[15], it was observed that all of them displayed high rates of spontaneous tumorigenesis. Despite the inherent variations associated with this approach, it is important to note that a common feature of the tumorigenic process observed in these mice models is that the tumors are always developed at old ages, which suggests that aneuploidy itself may be a promoting factor rather than a tumor initiator. However, the involvement of MAD2 on therapy response is still unclear, since some data suggest that over expression of MAD2 increases both sensitivity and resistance to a specific therapy.
In vitro analyses of tumor cell lines have shown that MAD2 levels influence chemotherapy response. Low levels of MAD2 increase resistance to platinum compounds, vincristine and γ- irradiation both in nasopharyngeal and Testicular Germ Cellular Tumor (TGCT) cancer cells[16,17]. In ovarian and gastric cancer cell lines, it has been demonstrated that through modulation of MAD2 levels using specific shRNAs, expression of p31comet[18] or micro-RNAs, cells become more resistant to PTX[19-22]. Resistance to PTX seems to be correlated with apoptosis inhibition or senescence induction[19]. This evidence suggests that a nonfunctional SAC caused by insufficient kinetochore proteins would not be able to activate the apoptotic pathway. Moreover, we have also demonstrated that in MKN45 gastric cancer cells, reducing MAD2 protein levels confers resistance to PTX. Even though we did not observe any modifications in apoptotic induction pathways, we did note a significant induction of senescence accompanied by an elevation in IL 6 and IL-8 levels, of a consequence of MAD2 downregulation. (Figure 2)
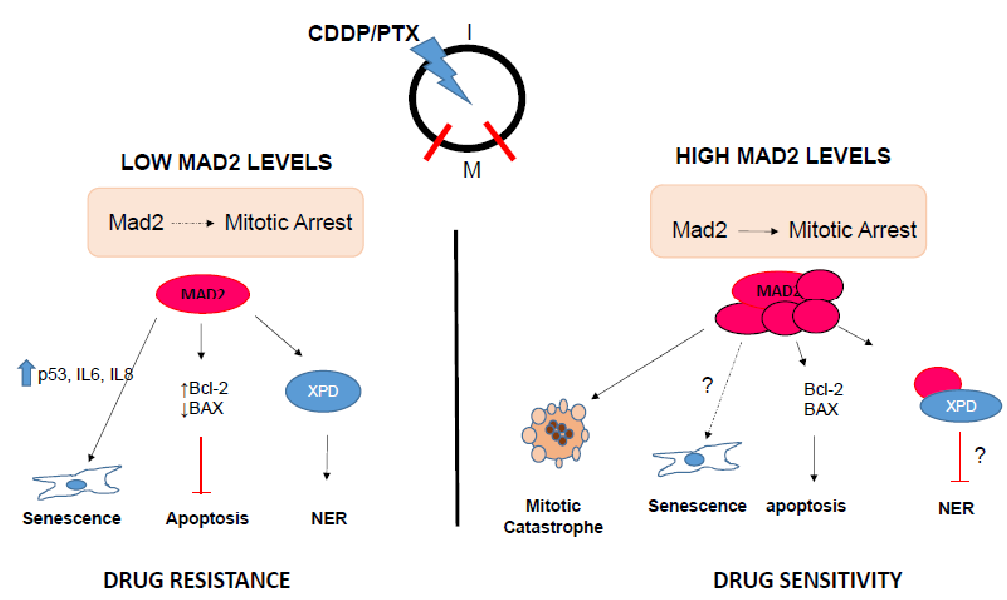
Figure 2:
It is important to be aware of the fact that the in vitro cellular systems do not always mimic the cellular processes that occur in vivo; tumors are subjected to multifactorial changes and undergo dramatically different developments between patients and therefore the expected response to a specific drug is not universal. Up until now, the available studies on MAD2 expression and chemotherapy response in patients have often been controversial. In general, taking into account the most relevant clinical studies, we can infer that elevated levels of MAD2 correlate with poor prognosis, evidenced by lower overall survival (OS) and or PFS (table 1). In contrast, immunohistochemical analysis of MAD2 within a series of high-grade serous epithelial ovarian cancer cell lines from patients who had previously been treated with CDDP and/or PTX, showed that low level MAD2 expressing patients had a shorter survival period and slower tumor progression[23]. It has also been shown that elevated MAD2 expression may lead to resistance to CDDP in advanced uterine cervical cancer patients, which is in clear disagreement with what seems to happen in the available in vitro studies with PTX[24]. However, we should take into account the following factors: On one hand, the criteria for gradation of MAD2 levels are not adequately standardized. This could explain the differences between the conclusions of the available studies. On the other hand, the cell type or the nature of the tissues analyzed in these previous studies should be taken as well in consideration.
Table 1:
| TUMOR | MAD2 LEVELS | CLINICAL PARAMETERS | REFERENCES |
|---|---|---|---|
| Ovary | HIGH | tPFS | Furlong, Fitzpatrick et al., 2012 |
| LOW | tPFS | McGrogan B. et al. 2014 | |
| Colon | LOW | tos | Li y Zhang , 2004 |
| Lung | LOW | tos | Sotillo , Schvartzman et al., 2010 |
| HIGH | tOS/WFS | Tatsuya Kato et al. 2011 | |
| Urothelial Bladder | HIGH | toS | Choi JW et al, 2013 |
| Oral | HIGH | toS | Teixeira et al.,2015 |
| Endometria l | HIGH | toS | Lin Li et al., 2013 |
| Neuroblastoma | HIGH | toS | Kohei Otake et al., 2011 |
After careful analysis of all available data, we propose a potential model where MAD2 protein levels on the primary tumor, would decide the effect of the given treatment (Figure 3). In resectable primary tumors with low MAD2 expression, surgery followed by adjuvant chemotherapy continues to be the only curative option and presents a better outcome. However, in some cases, the tumor relapses due to resistance to apoptosis and/or senescent phenotype induction in these conditions, which can complicate the clinical landscape. On the contrary, resectable primary tumors with high MAD2 show a better clinical outcome after adjuvant therapy, suggesting that MAD2 is required for apoptosis induction in primary tumors diagnosed at early stages. However, in advanced stage tumors with high levels of MAD2, presenting metastasis and therefore poor prognosis, treatment is inefficient and our model suggests that downregulation of MAD2 could improve clinical outcome in this particular situation. (Figure 3)
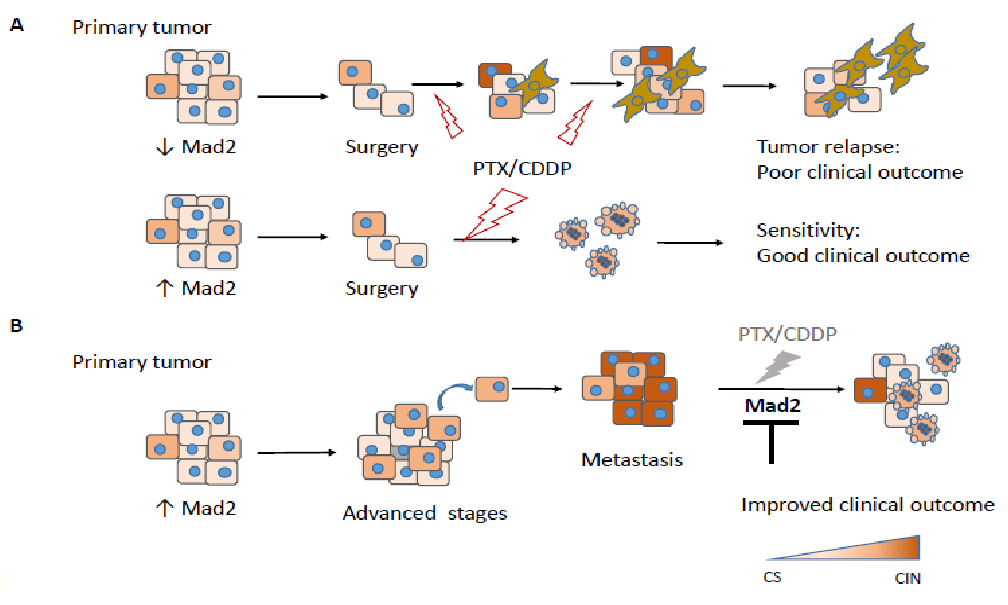
Figure 3:
The results available in the literature indicate that, although it is hard to predict the unique responses of a patient to therapy based on its levels of expression of MAD2 at the time of diagnosis, it is not clear whether altered MAD2 levels do affect therapy response, as this seems to depend on the origin of the tumor.
MAD2, more than a mitotic regulator?
The above data indicate that MAD2 is acting as an Oncogene, and could be used as a prognosis marker. However, the role of MAD2 in response to therapy is unknown, since the data available from the literature are contradictory and the majority of them are based on in vitro systems. MAD2 seems to be involved in various functions along the cell cycle, one of them being well characterized and specific to mitosis, and another one which is involved in the control of the interphase. Additional functions should be investigated, in relation with the control of cell fate after cellular damage, and also in the context of cancer therapy response.
The first evidence of MAD2 as a modulator of DNA damage response was obtained using Hela Cells, in a set of experiments where the authors demonstrated that the delay in mitosis induced by DNA damage is not due to an ATM-mediated DNA damage checkpoint pathway[25]. Other groups observed an interplay between ATM and SAC activation[26]. A novel connection between both processes has been described, where a physical interaction between nuclear MAD2 and Chk1 was detected in stress-free conditions, being this interaction stronger when cells presented DNA damage. One possibility to explain these findings is to consider that MAD2 plays a role controlling the translocation of DNA damage repair proteins to the nucleus[27]. In agreement with these results, MAD2 has been shown to be localized at the nuclear pore during interphase and to interact with TRp1 and Nup153 proteins[28]. For instance, in a work performed using HeLa cells, it was demonstrated that MAD2 interacts with XPD and ERCC1, two key proteins that participate in nucleotide excision repair pathway (NER). It was shown that in MAD2 overexpressing cells, XPD was localized in the cytoplasm and its recruitment to the nucleus after CDDP treatment was weak, whereas XPA was over expressed and accumulated in the nucleus. Some authors suggest that high levels of MAD2 can sensitize to CDDP through its ability to reorganize repair proteins, which could play a role sequestering XPD in the cytoplasm and therefore making the NER pathway weaker or inefficient. On the other hand, they did not find any relationship between MAD2 and Rad51, which are both involved in homologous recombination repair[24].
Other observations suggest a role for MAD2 within the apoptosis signaling pathway, for instance MAD2 downregulation correlates with a reduction on the expression of apoptotic proteins and on caspases activity within the mitotic cell population[29]. Supporting these data, in the germ tumor cell line (GCT) 1411HP, low levels of MAD2 correlate with an inhibition of the MEK/ERK and apoptotic pathways[17]. Furthermore, downregulation of MAD2 in GC and nasopharyngeal tumor cell lines increased cisplatin resistance due to the fact that the ratio of anti-apoptotic protein Bcl-2 levels and pro-apoptotic protein Bax was increased, whereas apoptosis was related to the expression of proteins such as cytochrome c, while cleaved caspase 3 maintained low levels[29,30].
The first observation that correlates MAD2 with senescence, came from a study in which low doses of Doxorrubicine induced senescence[31]. In other studies, depletion of MAD2 in human fibroblast cell line (IMR-90) confirmed the induction of senescence through the p53 signaling pathway[32]. These results suggest that aneuploidy induced by MAD2 depletion, is sensed as a stress signal by normal cells, which in turn will trigger premature cellular senescence that acts as a barrier to cell proliferation in cells with unbalanced karyotypes, and not related with DNA damage. Supporting this data it has also been proved that overexpressing p31comet, which acts as a spindle checkpoint inhibitor, inducing tumor cell senescence by mediating accumulation of p21 (Waf1/Cip1) and MAD2 disruption[33]. In addition, our data corroborate this fact[20].
Conclusions and Future Perspectives
In this review, we have highlighted the relevant evidence of the implication of MAD2 protein levels as a marker which is in fact capable to define patients overall survival. To sum up, upregulation of MAD2 protein levels correlates with low survival, indicating that MAD2 is a good prognosis biomarker. It is clear that MAD2 plays other functions beyond mitosis; however further studies need to be done to clarify the partners and the real function of MAD2, since at the moment only in vitro correlations exist between survival /dead and MAD2 levels. There is a lack of in depth studies characterizing the molecular mechanism involved in this processes. The fact that MAD2 regulates the response to therapy, suggests that it could be a new therapeutic target. However, some concerns should be raised, in order to facilitate the development of drugs targeting MAD2. These doubts come from the in vitro analysis, where depletion of MAD2 confers senescence-like phenotype after treatment with agents used routinely in cancer therapy. Sadly there are no clinical trials corroborating the effect of MAD2 function in patients. However, the silencing MAD2 expression using siRNAs, increases sensitivity to chitosan in non-small cell lung model[34,35]. In addition, new synthetic antimitotic drugs showed that its main apoptotic potential is due to its ability to disrupt the SAC machinery and therefore provoking a mitotic catastrophe[36].
There are many gaps to fill in the current knowledge of the processes regulating cell fate, so future studies will need to answer some of this crucial question.
How is MAD2 regulated? Is it really the amount of protein what makes the difference or is it its availability to be in the right place and the right conformation state? And, is this balanced by its interactions with other SAC proteins?
Does MAD2 predict the potential response to therapy, or is it on the other hand specific for a certain drug and/or a certain type of tumor?
How do MAD2 levels influence the other mitotic complex proteins or its functionality?
Acknowledgement:
This work was supported by a grant from UAM-SANTANDER CEAL-AL/2013-29. JBI was supported by a fellowship from Catedra Isaac Costero, funded by Banco Santander-UAM and is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received fellowship CVU:607546 from CONACYT.
Conflict of Interest:
The authors declare that they have no competing relationship or commercial affiliations or any financial interests.
Abbreviations: SAC: Spindle Assembly Checkpoint; CIN: Chromosome Instability
References
- 1. Musacchio, A., Salmon, E.D. The spindle‐assembly checkpoint in space and time. (2007) Nat Rev Mol cell Biology 8(5): 379-393.
- 2. Lara-Gonzalez, P., Westhorpe, F.G., Taylor, S.S. The spindle assembly checkpoint. (2012) Curr Biol 22(22): R966-R980.
- 3. Jia, L., Kim, S., Yu, H. Tracking spindle checkpoint signals from kinetochores to APC/C. (2013) Trends Biochem Sci 38(6): 302-311.
- 4. Chang, L., Barford, D. Insights into the anaphase-promoting complex: a molecular machine that regulates mitosis. (2014) Curr Opin Struct Biol 29:1-9.
- 5. Ballister, E.R., Lampson, M.A. Chromosomal Instability: Mad2 beyond the spindle checkpoint. (2012) Curr Biol 22(7): R233-R235.
- 6. Luo, X., Tang, Z., Xia, G., et al. The Mad2 spindle checkpoint protein has two distinct natively folded states. (2004) Nat Struct Mol Biol 11(4): 338-345.
- 7. Luo, X., Tang, Z., Rizo, J., et al. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. (2002) Mol cell 9(1): 59-71.
- 8. Luo, X., Fang, G., Coldiron, M., et al. Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. (2000) Nat Struct Mol Biol 7(3): 224-229.
- 9. Westhorpe, F.G., Tighe, A., Lara-Gonzalez, P., et al. p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. (2011) J cell sci 124(Pt 22): 3905-3916.
- 10. Habu, T., Matsumoto, T. p31(comet) inactivates the chemically induced Mad2- dependent spindle assembly checkpoint and leads to resistance to anti-mitotic drugs. (2013) SpringerPlus 2: 562.
- 11. Xia, G., Luo, X., Habu, T., et al. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. (2004) EMBO J 23(15): 3133-3143.
- 12. Kato, T., Daigo, Y., Aragaki, M., et al. Overexpression of MAD2 predicts clinical outcome in primary lung cancer patients. (2011) Lung Cancer 74(1): 124-131.
- 13. Zhang, S.H., Xu, A.M., Chen, X.F., et al. Clinicopathologic significance of mitotic arrest defective protein 2 overexpression in hepatocellular carcinoma. (2008) Hum Pathol 39(12): 1827-1834.
- 14. Li, M., Zhang, P. Spindle assembly checkpoint, aneuploidy and tumorigenesis. (2009) Cell cycle 8(21): 3440.
- 15. Sotillo, R., Hernando, E., Diaz-Rodriguez, E., et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. (2007) Cancer Cell 11(1): 9-23.
- 16. Wang, X., Jin, D.Y., Wong, Y.C., et al. Correlation of defective mitotic checkpoint with aberrantly reduced expression of MAD2 protein in nasopharyngeal carcinoma cells. (2000) Carcinogenesis 21(12): 2293-2297.
- 17. Fung, M.K., Cheung, H.W., Ling, M.T., et al. Role of MEK/ERK pathway in the MAD2-mediated cisplatin sensitivity in testicular germ cell tumour cells. (2006) Br J cancer 95(4): 475-484.
- 18. Date, D.A., Burrows, A.C., Venere, M., et al. Coordinated regulation of p31(Comet) and Mad2 expression is required for cellular proliferation. (2013) Cell cycle 12(24): 3824-3832.
- 19. Hao, X., Zhou, Z., Ye, S., et al. Effect of Mad2 on paclitaxel-induced cell death in ovarian cancer cells. (2010) J Huazhong Univ Sci Technolog Med Sci 30(5): 620-625.
- 20. Bargiela-Iparraguirre, J., Prado-Marchal, L., Pajuelo-Lozano, N., et al. Mad2 and BubR1 modulates tumourigenesis and paclitaxel response in MKN45 gastric cancer cells. (2014) Cell cycle 13(22): 3590-3601.
- 21. Otake, K., Uchida, K., Tanaka, K., et al. HsMAD2 mRNA expression may be a predictor of sensitivity to paclitaxel and survival in neuroblastoma. (2011) Pediatr Surg Int 27(2): 217-223.
- 22. Tambe, M., Pruikkonen, S., Maki-Jouppila, J., et al. Novel Mad2-targeting miR-493-3p controls mitotic fidelity and cancer cells' sensitivity to paclitaxel. (2016) Oncotarget 7(11): 12267-12285.
- 23. Furlong, F., Fitzpatrick, P., O'Toole, S., et al. Low MAD2 expression levels associate with reduced progression-free survival in patients with high-grade serous epithelial ovarian cancer. (2012) J Pathol 226(5): 746-755.
- 24. Morishita, M., Sumi, T., Nakano, Y., et al. Expression of mitotic-arrest deficiency 2 predicts the efficacy of neoadjuvant chemotherapy for locally advanced uterine cervical cancer. (2012) Exp Ther Med 3(2): 341-346.
- 25. Mikhailov, A., Cole, R.W., Rieder, C.L. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. (2002) Curr Biol 12(21): 1797-1806.
- 26. Eliezer, Y., Argaman, L., Kornowski, M., et al. Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. (2014) J Biol Chem 289(12): 8182-8193.
- 27. Chila, R., Celenza, C., Lupi, M., et al. Chk1-Mad2 interaction: a crosslink between the DNA damage checkpoint and the mitotic spindle checkpoint. (2013) Cell cycle 12(7): 1083-1090.
- 28. Mossaid, I., Fahrenkrog, B. Complex Commingling: Nucleoporins and the Spindle Assembly Checkpoint. (2015) Cells 4(4): 706-725.
- 29. Du, Y., Yin, F., Liu, C., et al. Depression of MAD2 inhibits apoptosis of gastric cancer cells by upregulating Bcl-2 and interfering mitochondrion pathway. (2006) Biochem Biophys Res Commun 345(3): 1092-1098.
- 30. Cheung, H.W., Jin, D.Y., Ling, M.T., et al. Mitotic arrest deficient 2 expression induces chemosensitization to a DNA-damaging agent, cisplatin, in nasopharyngeal carcinoma cells. (2005) Cancer Res 65(4): 1450-1458.
- 31. Eom, Y.W., Kim, M.A., Park, S.S., et al. Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. (2005) Oncogene 24(30): 4765-4777.
- 32. Lentini, L., Barra, V., Schillaci, T., et al. MAD2 depletion triggers premature cellular senescence in human primary fibroblasts by activating a p53 pathway preventing aneuploid cells propagation. (2012) J Cell Physiol 227(9): 3324-3332.
- 33. Yun, M., Han, Y.H., Yoon, S.H., et al. p31comet Induces cellular senescence through p21 accumulation and Mad2 disruption. (2009) Mol Cancer Res 7(3): 371-382.
- 34. Nascimento, A.V., Singh, A., Bousbaa, H., et al. Mad2 checkpoint gene silencing using epidermal growth factor receptor-targeted chitosan nanoparticles in non‐small cell lung cancer model. (2014) Mol Pharm 11(10): 3515-3527.
- 35. Nascimento, A.V., Gattacceca, F., Singh, A., et al. Biodistribution and pharmacokinetics of Mad2 siRNA-loaded EGFR-targeted chitosan nanoparticles in cisplatin sensitive and resistant lung cancer models. (2016) Nanomedicine (Lond) 11(7): 767-781.
- 36. Masawang, K., Pedro, M., Cidade, H., et al. Evaluation of 2',4'-dihydroxy-3,4,5-trimethoxychalcone as antimitotic agent that induces mitotic catastrophe in MCF-7 breast cancer cells. (2014) Toxicol Lett 229(2): 393-401.










