
Molecular Mechanisms of Drug Abuse, Dependency and Craving
Joseph D. Miller2
Affiliation
- 1Division of Pharmacology and Toxicology, College of Pharmacy, The University of Texas at Austin, Austin TX 78712, USA
- 2Department of Pharmacology, American University of the Caribbean School of Medicine, Cupecoy, St. Maarten
Corresponding Author
Richard E. Wilcox, Ph.D, Professor of Neuropharmacology, University of Texas, College of Pharmacy 2409, University Avenue STOP A1900 Austin, TX 78712-1113; Tel: +(512) 471-5199; E-mail: wilcoxrich@austin.utexas.edu
Citation
Wilcox, R.E., et al. Molecular Mechanisms of Drug Abuse, Dependency and Craving. (2016) J Addict Depend 2(1): 1- 12.
Copy rights
© 2016 Wilcox, R.E. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Drug abuse; Drug dependence; Addiction; Substance use disorder; Genetics of drug dependence; Theories of drug dependence; Treatment of drug dependence;Drugs for craving
Introduction
How serious are drug abuse and drug dependence? Currently in the USA, alcohol abuse is associated with an overall cost of $224 billion, tobacco abuse, $295 billion and illicit drug abuse, $193 billion[1]. How many people are affected? As an example in the USA, among people ages 18 or older, the lifetime prevalence of alcohol use was 87%, while 25% of people reported binge drinking in the past month (in 2013)[2,3]. Similarly, in the USA, 7% of people ages 18 and older (17 million adults) had an Alcohol Use Disorder (AUD)[3].
Most people who become serious substance users (DSM-V; drug dependent in DSM-IV) initially are drug abusers. Many drug abusers never go on to develop drug dependence. The essential difference between drug abuse and drug dependence is that the latter is a medical disease characterized by a loss of control over drug use and drug seeking (DSM-IV, DSM-V[4,5]).
The above suggests and the literature has demonstrated that clinically there are two sub-populations of people who use drugs: drug abusers and drug dependent people. Drug abuse is under voluntary control and can be reduced via coercion, education, reduced drug supply, punishment, and increased drug cost[5]. In contrast drug dependence (severe drug use disorder) cannot be reduced by these means and is associated with loss of control over drug seeking and taking and a marked craving for the drug (to the exclusion of other thoughts and activities)[6].
A large body of evidence now shows that drug dependence is much more likely to occur with drug exposure in at risk individuals - people with specific genetic deficits associated with impaired neural activity in the brain’s pleasure pathway (dopaminergic projection from the ventral tegmental area of Tsai, VTA) and its projections to the limbic system (especially the nucleus accumbens, frontal and prefrontal cortices)[4,7].
Nature and Mechanisms of Craving
Drug dependence develops as an “adaptive” process in which the firing of neural circuits associated with emotion generally and stimulus salience and reward more specifically is altered long-term[7,8,9]. As such, drug dependence parallels other types of learning in many ways.
An important finding in the addiction research field is that, although the mechanisms of action of addictive drugs differ substantially, they are all able to alter the activity of a “final common pathway” running from the VTA to the nucleus accumbens and the frontal/prefrontal cortices[4,7]. This represents a challenge and also an opportunity for individuals who wish to treat drug dependence. The challenge is that there are many ways (neural mechanisms) by means of which the activity of this pathway can be altered (e.g., ethanol does not have the mechanism of action of cocaine but both can change the firing of the VTA pathway). The opportunity is that addiction theories and observations of currently effective anti-craving drugs support the idea that altering the activity of the VTA pathway in an appropriate direction may reduce drug craving[7].
Brief overview of Addiction theories
Elsewhere, we have recently provided a thorough comparison of some of the most significant theories of the development and maintenance of drug dependence[7]. In the present context our focus will be on those aspects of theory that most directly lead to ideas about specific drug MOA that are likely to be involved in reversing the “addictive process” and the reduction of drug craving. Briefly, the addictive process may be viewed as having the components of LIKE, WANT and NEED, each of which plays a role in the development and maintenance of drug taking/drug seeking and craving (see Figure 1).
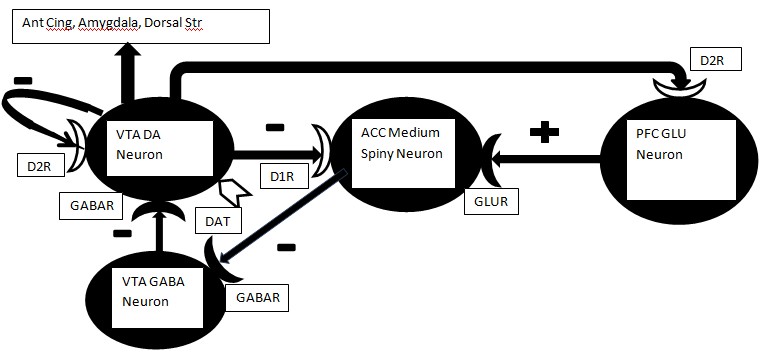
Figure 1: Down-regulation of presynaptic or postsynaptic DA receptors on the DA VTA neuron, medium spiny neuron or Glu neuron in mPFC will result in elevated DA release. Any reduction in the activity of DAT should yield a similar elevation of DA in the synaptic cleft. This elevation may be the neurochemical correlate of “want.” However, as down-regulation proceeds it is possible that the DA neurons will be largely depleted of releasable DA and any further application of psychostimulant drugs may result in diminishing returns, i.e., further down-regulation of DA receptors and associated depletion of DA stores in VTA neurons. White coloration indicates potential sites of DA receptor down-regulation or reduced DAT activity.
Abbreviations: VTA = ventral tegmental area, D1R = Dopamine D1-like receptor, D2R = Dopamine D2-like receptor, GABAR = GABA A receptor, DAT = Dopamine transporter, ACC = nucleus accumbens, GLUR = Glutamate receptor, PFC = medial prefrontal cortex, Ant Cing = anterior cingulate cortex, Dorsal Str = dorsal striatum.
LIKE refers to the euphoria associated with drug use (especially drug use in the early stages of drug exposure)[8,10-13]. In animal models, LIKE is synonymous with positive reinforcement, the neural mechanism of which is the activation of dopamine neurons in the VTA and the release of dopamine in the nucleus accumbens.
WANT occurs once the addictive process is underway and is associated with drug craving as the person’s limbic system functions “normally” only in the presence of the drug[9,14-16]. A positive feedback loop occurs in which the more drug that is consumed, the more that drug is desired. Thus, a progressively greater percentage of the person’s time is spent in drug seeking behavior. For psycho-stimulant drugs such as cocaine and the amphetamines, this phase of the addictive process is associated with a behavioral sensitization (enhanced response with repeated exposure to the drug) to the stimulant drug[10-13]. As shown in Figure 1, the continued use of amphetamine will down-regulate the D1 receptor on MSN (medium spiny) neurons and also the D2 receptor on the glutamate cortical projection to MSN. The result is less inhibition of the MSN neuron and a greater excitatory input from cortex. Thus, the MSN neuron fires and inhibits the local GABA neurons in the VTA. This action in turn disinhibits the DA neuron in the VTA. DA target neurons outside the striatum get a massive release of DA which is the neural substrate of behavioral sensitization[7].
Once the addictive process is well developed, the NEED condition manifests. Here, the person’s brain is so well adapted to the drug’s presence that the absence of the drug is associated with intense cravings and strongly negative emotional states[17-20]. At this stage, the person’s brain exhibits widespread down-regulation of DA receptors beyond the striatum and the “vicious cycle” of accelerated drug use. This neurochemical mechanism is further strengthened by conditioning to secondary reinforces associated with drug use (environmental cues), yielding craving. Addiction theory development has been somewhat “uneven” in that those investigators who have provided the greatest amount of information on brain changes associated with WANT have tended to focus their efforts on the earlier phases of the addictive process whereas those investigators who have provided the greatest amount of information on brain changes associated with NEED have tended to focus their efforts on the later phases of the process[7,9,16]. However, recent integrative formulations[7,9] have shown that these differing emphases can be resolved without weakening the overall theoretical structure. What is clear from the above discussion is that there are phases in the development of drug dependence that may represent distinguishable patterns of neural activity. In parallel to the physiological studies of the neural substrates of drug dependence, neurochemical studies have likewise demonstrated phases in dependence development. Together, these studies suggest that anti-craving treatment approaches may have viable and discernible targets.
Anti-craving drug mechanisms
In this section we will review each neurochemical component of the drug dependence pathways in relation to agents used in an attempt to reduce drug craving.
Dopaminergic Agents (DA): Because of the extensive work showing the relevance of the VTA pathway to drug dependence and the fact that psycho-stimulants (such as cocaine and the amphetamines) appear to act directly on the DA neuron, a considerable effort has been expended in evaluating the potential of drugs acting via dopamine to alter craving[17,21-27].
Dursteler, et al.[21] reviewed studies in which methylphenidate (MPH, a dopamine uptake inhibitor used in treating attention deficit hyperactivity disorder, ADHD) could be used as a cocaine substitute to reduce cocaine craving. To date, the results overall were disappointing when MPH was used in persons who did not also have a diagnosis of ADHD. One potential reason for the treatment failure is that MPH is an extremely effective blocker of DA uptake. On the other hand, what is needed for such “substitution” therapy is an agent that only weakly blocks the dopamine transporter (such as modafinil) or an agent that combines blockade of the DA transporter with other actions (such as blocking the norepinephrine and dopamine transporters by bupropion). Below we will provide evidence to substantiate this position.
O’Brien’s[28] review was one of the early reports of the effectiveness of bupropion on alleviating the craving for nicotine. Islam and Rahman[29] have discussed the role of bupropion as a part of the treatment of the craving for nicotine. Modafinil is an agent primarily used to reduce sleepiness in persons suffering from jet lag or narcolepsy. Its mechanisms of action include a modest direct stimulation of alpha-noradrenergic receptors and, significantly, a weak blockade of DA transport. Modafinil appears to reduce craving to cocaine. This is consistent with the view that an ideal agent for “substitution” therapy would have some ability to mimic the addictive drug but do so more weakly (have reduced pharmacological efficacy compared to the addictive agent). Essentially, an ideal therapeutic agent for carving would not allow the person to get “high” but would still be able to reduce craving by weakly mimicking the addictive agent’s actions.
George, et al.[22] evaluated the roles of dopamine and Corticotropin Releasing Factor (CRF) in the neurochemical mechanism of craving. They postulated two types of brain activities. The within-system process is one in which the addictive agent elicits an opposing (“neutralizing”) reaction in the same system where the drug has its major action. The author’s between- system process is one in which the addictive drug recruits brain circuits beyond the one in which it has its primary effect. The authors postulated that repeated reduced dopaminergic activity coupled with enhanced activity in the CRF system (during drug withdrawal) is a critical part of the mechanism of dependence (we would say especially in development of NEED). In 2013 Koob[17] suggested that compulsive drug seeking in alcoholism occurs via negative reinforcement (an action that reduces a negative emotional state, i.e., withdrawal). He argued that in addition to dopamine and CRF other modulators of the brain stress response including norepinephrine, dynorphin, and neuropeptide Y could play a role in the extended amygdala (a brain region linking emotion and memory) in mediating the drug seeking that attempts to reduce the reward deficit in the dependent brain.
Lee, et al.[30] reviewed studies indicating some conflict between the results of human trials and tests of therapy in animal models of psycho-stimulant abuse. These authors highlighted a need for drug combination therapy used in a specific temporal sequence. Thus, a dopamine agonist (that would mimic some aspects of DA itself at one of DA’s receptors) would be given initially followed by a second agent (such as a serotonin or neurokinin-1 receptor antagonist) for normalizing behavior. Myers[23] noted that amperozide (a serotonin-2 receptor antagonist that also releases dopamine) is able to attenuate alcohol craving in animal models. Similarly, Nava, et al.[24] found that gamma-hydroxybutyrate[1], naltrexone, or disulfiram were all equally able to reduce both craving for alcohol and biological markers of alcohol abuse in alcoholics. GHB inhibits dopamine release; naltrexone blocks opioid receptors and indirectly modulates DA neural activity in some brain regions; and disulfiram inhibits ethanol metabolism but may also reduce the metabolism of dopamine via MAO (Monoamine Oxidase).
In contrast, Pierce, et al.[25] noted that while effective therapies for reducing craving for alcohol, opioids and nicotine have emerged, promising medications to reduce craving for psycho-stimulants have not fared well in clinical trials. One possible reason for this, highlighted by Lee above[30], is that a combination of agents (or agents given in a specific temporal sequence) may be needed to fully reverse the neural adaptations associated with craving, in order to manifest a beneficial effect. Thus, Rothman, et al.[26] emphasized the value of dual dopamine/serotonin releasers as treatments for psycho-stimulant addiction. The efficacy of such an agent suggests that serotonin release may dampen the dopamine-mediated stimulant effects yielding a benefit on craving without getting the person “high”. Finally, Self[27] summarized a large body of work on neurochemical adaptations that occur with chronic exposure to addictive drugs. He indicated that whereas dopamine triggers relapse by stimulating D2 DA receptors that inhibit cAMP, drugs that selectively activate D1 DA receptors (and enhance cAMP) prevent relapse and may be of benefit in reducing craving. Thus, the type of DA mimetic agent should be considered in anti-craving drug development.
Serotonergic (5HT) Agents: As indicated above, one of the roles played by 5HT within the limbic system is the modulation of dopamine activity (especially dopamine release). Ago, et al.[31] looked at psycho-stimulant induced behavioral sensitization in mice and its modulation by serotonergic agents. Serotonergic receptor blockers tend to have actions at a large number of serotonin receptors because of the marked similarities in affinities across the receptor family. In the Ago study, the authors found that osmemozotan (selective serotonin-1A agonist), ritanserin (a serotonin-2A/2C antagonist)1 , and azasetron (serotonin-3 antagonist) inhibited the development and maintenance of sensitization to varying extents with the 5HT3 antagonist having the greatest clinical effects. Johnson[32] likewise demonstrated that ondansetron (an antagonist of the 5HT3 receptor) was able to reduce craving significantly in a subpopulation of alcohol-dependent patients.
Hauser, et al.[33] examined the most recently discovered member of the 5HT receptor family, the 5HT7 receptor, since links have been shown between alcoholism and variants of the 5HT7 receptor gene. Blockade of the 5HT7 receptor with SB269, 970 prevented amphetamine-induced inhibition of neural firing in the VTA (cited in (33)), but the neuroanatomical mechanism whereby this effect was achieved was unclear. Lanteri, et al.[34] showed that chronic dosing with several different addictive drugs sensitized 5HT and noradrenergic (NE) neurons by disrupting the reciprocal inhibition between them. Furthermore, these authors demonstrated that RS10, 222 (5HT2C agonist) reversed the effects of the addictive drugs and the authors suggested that a 5HT/NE uncoupling is a common neurochemical consequence of repeated consumption of addictive agents. As discussed above, Lee, et al.[30] showed that sequential administration of a dopaminergic drug followed by a second agent (such as a 5HT2A/2C antagonist) that modified dopamine activity indirectly was beneficial in reducing psycho-stimulant induced sensitization. Also we noted above that Myers[23] reported that amperozide (a serotonin-2 receptor antagonist that also releases dopamine) attenuates alcohol craving in animal models. Finally, we also indicated above that Rothman, et al.[26] emphasized the potential value of dual dopamine/serotonin releasers as potential treatments for psycho-stimulant addiction due to a dampening effect of 5HT release on dopamine-mediated stimulant effects.
Noradrenergic Agents (NE): NE drugs are generally not as well recognized for their anti-craving potential as the DA and 5HT drugs discussed above. However, there is a substantial literature indicating a role for NE transmission in the induction of the addictive process and in the development and modulation of drug craving especially for nicotine. The effects of the dual action agent bupropion (that blocks both DA and NE transport) were discussed above[28,29]. Interactions between 5HT and NE neurons were mentioned above in the work by Lanteri, et al.[34] who showed that chronic dosing with several different addictive drugs sensitized 5HT and noradrenergic (NE) neurons by disrupting a reciprocal inhibitory link between them and that 5HT/NE uncoupling is a common neurochemical consequence of repeated consumption of addictive agents.
In 2014 Lin[35] reported on the two general strategies for treating craving – substitution therapy and anti-craving medications. The review included a discussion of the role of bupropion in treating the craving for nicotine (strong effect) but also in treating craving for food in binge eating and obesity (which shows significant overlap in brain substrates and neurochemistry with the drug addictions). As discussed above, Nava, et al.[24] found that 3 agents that directly or indirectly altered DA and / or NE, (GHB, naltrexone, or disulfiram) could both reduce ethanol craving and aid in maintaining abstinence (see also (35)). Sofuoglu and Sewell[36] described the critical role of NE in mediating many effects of psycho-stimulants, including sensitization and the reinstatement of drug seeking (following extinction and a priming drug exposure). These authors noted that disulfiram appears to block NE synthesis as well as potentially inhibiting its metabolism. The same authors noted that lofexidine, an alpha-2 adrenergic direct agonist, reduced the craving induced by stress and drug cues in cocaine users. Upadhyaya, et al.[37] provided a novel perspective on the role of NE in craving. They discussed the abuse potential of the drug atomoxetine (which selectively blocks NE uptake) but found that the literature does not support the idea that selective NE uptake inhibitors are addictive. Thus, a drug that that selectively blocks NE, but not DA, transport would probably not reduce craving either. Nevertheless a drug that reduces noradrenergic transmission, e.g. lofexidine (by stimulating pre-synaptic alpha-2 axon terminal autoreceptors), may be useful in treating craving.
Cholinergic Agents: As presented above, Islam and Rahman[29] evaluated the effectiveness of treatments for nicotine craving. In addition to bupropion (DA and NE uptake inhibitor) they also evaluated varenicline, a nicotinic partial agonist (substitution therapy). The authors indicated that oral therapy with either agent had limitations. Similarly, O’Brien[28], in studying the same two agents, reported limitations in their effectiveness in reducing nicotine craving.
GABAergic Agents: As the major inhibitory transmitter in the brain, GABA should play some role in the addictive process and craving. Furthermore, some of the addictive drugs, in particular ethanol, have important direct actions on GABA receptors. Fehr, et al.[38] evaluated the effects of tiagabine (GABA uptake blocker) on the activation of the VTA pathway. The paradigm employed an intravenous challenge dose of ethanol in non-addicted people to determine if tiagabine attenuated the acute response to ethanol. This work hypothesized that stimulation of DA release in the limbic system by addictive agents could be reduced by augmenting GABA transmission. The author’s results using PET imaging did not support the idea that potentiating GABA synaptic levels (via blocking GABA transport) could diminish the effects of ethanol challenge on brain activity. A caveat in this study of course is that addicted people and normal individuals likely have differences in brain function. Also, the study is important in highlighting the anti-craving potential of the large set of anti-seizure/mood stabilizing agents now available clinically (see below). Consistent with the importance of the role of GABA in reducing craving. baclofen is a GABA-B direct agonist with a modest ability to reduce craving for alcohol and nicotine[35].
NMDA agents: Acamprosate was reported by Lin to have robust effects in reducing craving for ethanol, nicotine, opioids, and pathological gambling, with modest effects on amphetamine craving[35]. Acamprosate is an NMDA receptor partial co-agonist (binding to the glycine co-agonist site that must be occupied for the NMDA receptor-gated channel to open). Jung and Namkoong[39] emphasized that although acamprosate was effective, it worked well only in certain patients, suggesting a role for genotypic or phenotypic sub typing in the treatment of craving. Olive, et al.[40] recognized that, as the brain’s major excitatory transmitter, glutamate should logically play a role in the addictive process and craving. Furthermore, there is an extensive literature showing that addictive agents (including ethanol) alter glutamatergic function and that changes in brain glutamate activity modulate the VTA pathway[7].
Like acamprosate, D-cycloserine[2] acts as a co-agonist at the NMDA receptor glycine co-agonist site and facilitates the opening of the NMDA receptor channel[40]. DCS may reduce the craving for cocaine and for nicotine. Memantine is an interesting agent with its primary clinical use being symptom reduction in patients with dementia. This drug is a non-competitive NMDA receptor antagonist that blocks active NMDA receptors thereby preventing NMDA channel function. Since the activation of NMDA receptors is essential for encoding memory, the key to the clinical effectiveness of memantine in Alzheimer’s disease is its ability to target only those NMDA receptors that are active. In appropriate doses, memantine reduces excess stimulation of the receptors that would otherwise produce an “excitotoxic” destruction of neurons. Memantine may also block 5HT3 and nicotinic cholinergic receptors. Together, these mechanisms allow memantine to reduce craving to nicotine and heroin. However, the literature suggests that both glutamatergic agonists and antagonists are useful in treating craving, an unusual situation from a neuropharmacological perspective.
Also[35] N-acetylcysteine (NAC) may modulate both glutamate and DA by mechanisms other than its ability to facilitate the body’s antioxidant defense system. NAC appears able to somewhat reduce craving for cocaine (and for pathological gambling, a syndrome sharing much in common with the drug addictions in terms of changes in brain activity).
Opioid agents: Substitution therapy for opioids (initially heroin and now also prescription opioids) has been a mainstay of opioid addiction treatment for a generation. Initially, the direct opioid full agonist methadone was utilized[41]. Methadone fully activates the same opioid receptors that are activated by heroin. More recently, the advantages of administering partial opioid agonists either alone (buprenorphine) or combined with an opioid antagonist (naloxone) that was inactive orally but active when the drug combination was diverted for smoking or injection were discovered.
Fishman, et al.[42] described a woman dependent on opioids with co-morbid alcohol use disorder who was also depressed. While the case was complex and the patient’s progress was uneven, she eventually responded well to buprenorphine –naloxone plus her antidepressant. Grimm, et al.[43] evaluated the effects of the general opioid antagonist naloxone on sucrose craving in rats because of the relationship mentioned earlier between craving for drugs and for certain foods. Naloxone was somewhat effective in reducing the behavioral response for sucrose in protracted abstinence.
Koob[17] argued forcefully that alcoholism is a chronic emotional distress syndrome consistent with the activation of brain stress systems discussed in many of Koob’s reviews. Thus, involvement of the opioid system (including dynorphin) is expected. Consistent with this view, dynorphin (an endogenous opioid peptide that activates primarily kappa opioid receptors) levels are elevated after chronic dosing with psycho-stimulants or opioids[17].
By blocking opioid (especially mu) receptors with drugs such as naltrexone, nalmefene and naloxone, clinicians may disrupt the addictive process and begin to reinstate more normal patterns of neural firing within the VTA pathways. As discussed above[35], naltrexone is a competitive antagonist at mu and kappa opioid receptors. It is highly effective in preventing the euphoric effects of opioid agonists and can help improve abstinence. However, it is considered to be a second line agent (after methadone and buprenorphine) because of low patient adherence. This may be because naltrexone prevents endogenous opioids from activating opioid receptors whereas full or partial opioid agonists allow these receptors to be activated (by endorphins/enkephalins etc. as well as by the anti-craving drugs themselves) to at least some extent. For many drug dependent people, abstinence requires at least some activation of the relevant brain pathways rather than their complete inhibition! Nevertheless[24,28] investigators found that naltrexone was one of several therapeutic agents able to reduce craving in alcoholics and normalize at least some biological markers of alcohol use disorder.
Cannabinoid Agents: While marijuana use is widespread in the USA, marijuana dependence is considered to be less of a problem than dependence on heroin, nicotine, ethanol, and psycho- stimulants. Whereas the lifetime prevalence was 46% for adults ages 26 and older, daily use in the past month for adults ages 26 and older was 6%[44]. However, the role of the endogenous cannabinoid system in the abuse of or dependence on other agents may be significant. Gamaleddin, et al.[45] highlighted the role of the endogenous cannabinoid system in nicotine addiction. Rimonabant (SR141, 716) is a cannabinoid-1 (CB1) “inverse agonist/antagonist”. An inverse agonist is able to activate receptors in their “resting” state while a pure antagonist would merely prevent the endogenous transmitter from activating that receptor. Such an agent may be useful in the treatment of nicotine and other addictions. Yang, et al.[46] explored the potential role of the other major class of cannabinoid receptors (CB2). Both receptors are members of the GPCR (G protein coupled receptor) family; CB1 is primarily expressed in brain whereas CB2 resides primarily in peripheral tissues, especially those associated with immune system modulation. CB2 ligands may be of benefit as anti-craving drugs since they appear to be anti-convulsant (see below). As we have mentioned above and will discuss in detail in the final section of this paper, drugs that treat seizure disorders are able to do so because they restore the balance between excitatory and inhibitory activity in the brain, especially in the areas that constitute the epileptic focus. This action also leads naturally to the potential of these same drugs as mood stabilizers (for people with bipolar disorder) and also as anti-craving medications. Recent work has demonstrated that CB2 receptors are also present in brain, reinforcing their possible involvement in several CNS disorders.
CRF Agents: Koob and his collaborators have written extensively on the role of brain stress systems in the long-term, primarily negative, aspects of drug dependence and craving (NEED)[17,22]. Thus the role of CRF and the extended amygdala in stress and the view of addictions such as alcoholism as reward deficit disorders have helped to draw the attention of investigators to peptides in general and to CRF in particular. Much of the work done to date has focused on small molecules that can pass the blood -brain barrier or direct microinjection of peptides into the brains of animals. However, the continuing development of novel drug delivery systems that can allow drugs to cross the blood brain barrier (“e-cigarette” vaporization devices, fusion protein systems, etc.) is likely to speed this research. Koob summarized pre-clinical work showing that administration of a CRF antagonist can reduce drug self-administration in animals made dependent on cocaine, opioids, ethanol and nicotine[17].
Mood Stabilizers and Dopamine System Stabilizers
Over the last quarter century, it has been realized that successful treatment of epileptic seizures requires a reinstatement of the balance between brain excitation and brain inhibition and that any of a large variety of mechanisms can achieve this end (Table 1). In turn, investigators have similarly realized that the most effective treatment strategy for bipolar disorder (in which mood swings from depression to mania and back again) is to stabilize mood by using agents that can restore the balance between brain excitation and inhibition. Related to this is the idea that schizophrenia can be most effectively treated using novel (atypical) antipsychotic agents that block serotonin-2A receptors fully and D2 DA receptors partially or that act as partial DA and perhaps serotonin agonists (Table 2).
Table 1: Anti-seizure drugs
| GENERIC | MOA | TRADE |
|---|---|---|
| Sodium Channel Recovery | ||
| Phenytoin | Slows Na+ channel recovery | Dilantin |
| Carbamazepine | Slows Na+ channel recovery | Tegretol |
| Carbamazepine XR | same as carbamazepine | Carbatrol-Xr, Tegretolxr |
| Eslicarbazepine | Aptiom | |
| Lamotrigine | Slows Na+ channel recovery | Lamictal |
| Topiramate | Slows Na+ channel recovery | TOPAMAX, Trovendixr, Qudexyxr |
| Fosphenytoin | Phenytoin prodrug Slows Na+ channel recovery | Cerebryx |
| Oxcarbazepine | Slows Na+ channel recovery | Trileptil |
| Lacosamide | Increases SLOW inactivation of volt-gated Na channels | Vimpat |
| Rufinamide | Slows Na+ channel recovery | Banzel |
| Enhances GABA Activity | ||
| Lorazepam | GABA allosteric modifier at BDZ site* | Ativan |
| Clonazepam | GABA allosteric modifier at BDZ site | Klonopin |
| Diazepam | GABA allosteric modifier at BDZ site | Valium |
| Clobazam | GABA allosteric modifier at BDZ site | Anxibloc |
| Phenobarbital | GABA allosteric modifier at BDZ site** | Phenobarbitone |
| Primidone | Metabolized to phenobarbital | Mysoline |
| Valproic acid | Stimulates synthesis & Inhibits metabolism of GABA | Depakene |
| Gabapentin | Increases GABA release; Negative modulation of VGCC | Neurotonin |
| Tiagabine | Blocks GABA reuptake; Negative modulation of VGCC | Gabatril |
| Pregabalin | Increase GABA transport; Negative modulation of VGCC | Lyrica |
| Gamma-vinyl-GABA, VIGABATRIN | Inhibits GABA metabolism by blocking GABA-T irreversibly | Sabril |
| Reduces Glutamate activity | ||
| Felbamate | NMDA receptor BB at Glycine site | Felbatol |
| Propofol | NMDA receptor BB (& GABA-A SS) | Diprivan |
| PERAMPANEL | NON-COMPET AMPA Receptor BB | Fycompa |
| Calcium and Potassium channels | ||
| Ethosuximide | Reduces T-type Ca++ currents | Zarontin |
| Zonisamide | BB Na+ chan, BB T-type Ca++ chan, Increases GABA release | Zonegran |
| Ezogabine | Postasium channel modulator | Potiga |
| Somatic Vesicle Associated Protein | ||
| Levetiracetam | Modulates SVA2 | Keppra |
| Brivaracetam | Modulates SVA2 | |
ABBREVIATIONS
*BDZ is benzodiazepine; **BARB is barbiturate; SS = agonist; BB = blocker; SVA2 = synaptic vesicle associated protein #2 VGCC = voltage-gated calcium channels GTC = generalized tonic-clonic
Table 2: Antipsychotic Drugs
| GENERIC | MOA | TRADE |
|---|---|---|
| First generation antipsychotics [FGA] | ||
| chlorpromazine [LOW potency] | BB D2, mACh, alpha1 | THORAZINE |
| haloperidol [HIGH potency] [2D6,3A3/3A4] | BB mostly D2 DA receptors | HALDOL |
| perphenazine [MEDIUM potency] [2D6] | D2 BB | TRILAFON |
| loxapine [MEDIUM potency] | BB D2 DA, alpha1, mACH& 5HT | LOXATANE |
| fluphenazine [HIGH potency] | 5HT2A, D2 , alpha1, Histamine 1 BB | PROLIXIN |
| Second generation antipsychotics [SGA] | ||
| clozapine [1A2] | BB 5HT2A as well as D2 DA receptors, mACh, HIST1 BB | CLOZARIL, Versacloz |
| risperidone [2D6] | 5HT2A, D2 , alpha1 BB | RISPERDAL |
| paliperidone [9-hydroxyrisperidone] [2D6, 3A4 IN VITRO] | 5HT2A, D2 , alpha1 BB | INVEGA, depot is SUSTENNA |
| olanzapine [1A2] | 5HT2A, D2, mACh, Hist1, alpha1 BB | ZYPREXA |
| quetiapine [3A3/3A4] | 5HT2A, D2, alpha1, Hist1 BB | SEROQUEL |
| ziprasidone [3A3/3A4] | 5HT2A, D2, alpha1 BB, 5HT1A part ag | GEODON |
| iloperidone [2D6, 3A4] | 5HT2A, D2 , alpha1 BB | FANAPT |
| asenapine [1A2] | 5HT2A, D2 , alpha1, Histamine 1 BB | SAPHRIS |
| lurasidone [3A4] | 5HT2A BB, D2 BB. 5HT1A PA | LATUDA |
| SS = agonist; BB = blocker; part ag = partial agonist | ||
| DA SYSTEM STABILIZERS | ||
| arapiprazole [3AE/3A4] | D2 part ag, 5HT1A part ag. | ABILIFY |
| arapiprazoleXR | Abilify Maintena | |
| brexipiprazole [3A4, 2D6] | D2 & 5HT1A part ag. + antag at 5HT2A, alphaR, 5HT2B, 5HT7, &muscR | REXULTI |
It now appears almost axiomatic that mood-stabilizing drugs might have potential value as anti-craving agents. However, this idea was a long-time coming[4]. Initially, the leap was understanding that drugs used to restore a balance in brain activity between net excitation and net inhibition were of potential value in the treatment of epilepsy and a variety of drug mechanisms of action were identified in that context. Subsequently, the concept of restoring a balance between excitation and inhibition was extended to the treatment of manic episodes in bipolar patients and many antiepileptic agents have subsequently been found to be of value in treating this disorder as well. Finally, investigators realized that restoration of a balance in brain activity induced by such mood stabilizers could apply to the treatment of craving.
As noted above, topiramate is an anti-seizure agent and mood stabilizer (like tiagabine) that blunts alcohol craving. Its pharmacology is somewhat complex, including classic inhibition of the recovery of voltage-gated sodium channels (necessary for calcium influx and transmitter release) plus enhancing GABA activity at GABA-A receptors and blocking glutamate activity at AMPA/Kainate glutamate receptors. Olive reviewed a number of agents including the mood stabilizers gabapentin, lamotrigine, and topiramate[40]. Lamotrigine, gabapentin, and to some extent topiramate all block voltage-gated sodium and calcium channels. All three agents can reduce the craving for ethanol with some data suggesting that topiramate is superior to the “gold standard” naltrexone (see above). Lamotrigine also produces a beneficial reduction in craving for cocaine and for inhalants.
As with treatment of bipolar patients, treatment of psychosis has progressed from the simplistic use of drugs that blocked D2 DA receptors (reducing the positive but not the negative and cognitive symptoms of schizophrenia) to the use of atypical antipsychotic agents. Such agents exhibit a variety of MOA including blockade of 5HT2A receptors or acting as partial agonists at D2 DA receptors (see below). Not surprisingly, from the perspective of hindsight, many of the atypical “antipsychotic drugs” are of value as mood stabilizers and should be also considered as potential anti-craving agents.
Drugs that are partial D2 DA agonists have fairly recently found a place as atypical antipsychotics and subsequently as mood stabilizers. These drugs, including aripiprazole and brexipiprazole, are now called dopamine system stabilizers and fulfill the criteria that seem to be necessary for reducing craving as follows. We will use the example of how these drugs work in treating the symptoms of schizophrenia to illustrate the point. Schizophrenia is a disorder in which the primary deficit is an underactive frontal/prefrontal cortex. Thus, a person suffering from schizophrenia may exhibit negative and cognitive symptoms due to the direct effects of the disease on the frontal/prefrontal cortex and benefit from treatments that enhance DA in these brain areas. Simultaneously, the schizophrenic person may exhibit positive symptoms due to the release of subcortical areas from inhibition with the associated enhanced release of DA. These symptoms would benefit from a net reduction in DA stimulation of its receptors. DA system stabilizing agents such as aripiprazole fulfill both needs. Such drugs substitute for DA in the frontal and prefrontal cortex where it is deficient but displace DA within subcortical brain regions where it is in excess.
Similarly, a drug-dependent person has a net increase in activation of brain DA within the limbic system due to the direct or indirect actions of the addictive drug with acute exposure (LIKE) and during the early phases of the addictive process (WANT). If the DA is severely reduced, as during drug withdrawal and perhaps in the later phases of the addictive process (NEED), then craving ensues. However, if the person is given aripiprazole, the net stimulation of DA receptors is normalized cortical deficits reduced and subcortical excesses masked.
Other Types of Agents
In a 2013 review, Koob[17] noted that the brain stress systems mediated by CRF in the extended amygdala and the Hypothalamic-Pituitary-Adrenal (HPA) axis are dysregulated by all of the major drugs with addictive potential. As a group, the addictive drugs elevate adrenocorticotropic hormone (ACTH) and corticosterone systemically and CRF in the extended amygdala during their withdrawal after chronic dosing. One must be careful not to link withdrawal effects per se with craving since, while the processes may overlap, they also have distinct components[7]. As mentioned above[30] neurokinin-1 (NK-1) antagonists can reverse both the behavioral and neurochemical changes induced by psycho-stimulants in animals, particularly if the antagonist is given at a critical time following administration of a DA agonist (combination therapy). Very interestingly, Mahler, et al.[47] discussed the role of certain hypothalamic peptides called orexins. These peptides known as orexin-A and orexin-B play a role in sleep/wakefulness and the drug suvorexant is available as a competitive antagonist of oxexin A/B at both Ox1 and Ox2 receptors. The authors suggested that orexins may be involved in drug seeking that is triggered by external stimuli (including cues, contexts or stressors).
Potential Leads for Anti-Craving Agents
In this section we will highlight several productive strategies for development of improved treatment of drug craving. Overall, these approaches will either tend to stabilize the addictive process at its current level (substitution therapy) or reverse the process towards normal brain function (“reversal therapy”).
Substitution Therapy: Maintaining the addictive process at its current level for a given patient is a strategy that has worked extremely well for many years for people dependent on heroin. Methadone and subsequently buprenorphine (with or without naloxone) have allowed opioid-dependent people to function effectively without needing their “fix”. A key aspect of this therapy has been the use of an oral medication that activates the same receptors as the intravenous or smoked heroin (or oral prescription opioid agonist) mimicking the effects of the addictive drug in the brain. This allows sufficient receptor stimulation to prevent craving but it is hypothesized that the addictive process remains at its current level since the oral route of drug dosing (longer latency of drug onset) does not engage neural plasticity mechanisms to the same extent as those routes of administration (intravenous or inhalational) that lead to rapid increases (and decreases) in brain drug levels.
Buprenorphine, a partial agonist, activates opioid receptors at a lower pharmacological efficacy than that of full agonists such as heroin or methadone. This means that some receptor activation takes place but less than what occurs after heroin or methadone. The medication’s relative intrinsic efficacy must be sufficient high to prevent craving but ideally sufficiently low that the person does not get “high”. In addition, if a dependent individual attempts to increase the dose, buprenorphine then competes with heroin for the opioid receptors2, greatly reducing the addictive liability of the drug.
Similarly, substitution therapy to reduce nicotine craving using nicotine patches, nicotine gum, nicotine e-cigarettes etc. has been an effective development in this field. In treating nicotine craving it is important to note that many dependent people do need additional medications. These may include bupropion that potentiates both DA and NE via blocking their uptake into the nerve ending or the direct partial nicotinic agonist varenicline. Furthermore, nicotine from a cigarette passes the blood-brain barrier more rapidly than does intravenously administered nicotine. For that reason e-cigarettes appear to be a good substitution for cigarette smoking as well as an effective harm reduction strategy.
Substitution therapy has not really been attempted systematically for ethanol. Benzodiazepines only partially substitute and barbiturates are problematic. Similarly, substitution therapy has been quite disappointing for stimulants such as cocaine and the amphetamines. To reduce craving for stimulants, several direct or indirect DA agonists have been tried. These include direct D2 DA agonists, such as the anti-Parkinson’s disease agent bromocriptine, that produces significant nausea and amazingly has itself been abused by stimulant-dependent persons! In fact, most agents used in an attempt to reduce stimulant craving appear to be drugs of abuse themselves. Exceptions may include bupropion and modafinil (as discussed above). As a dual action drug that potentiates both DA and NE, bupropion may have less abuse liability because of the added increase in synaptic NE levels which may somewhat dampen some of the effects of DA. In contrast, methylphenidate (MPD) is a powerful inhibitor of the uptake of DA into the nerve ending used in children with ADHD to boost DA levels. Thus, MPD should be effective in reducing the craving for both cocaine and methamphetamine since it blocks the DA transporter and enhances synaptic cleft levels of DA, thereby mimicking the net effects of cocaine (transport blockade) and amphetamine (exchange diffusion, leading to enhanced DA release). Furthermore, MPD is a safe and highly effective treatment for ADHD in both children and adults. However, the fact that MPD shares a mechanism of action with cocaine is problematic in stimulant-dependent persons because it may induce a craving for itself! Thus, MPD has not worked well in clinical practice as an anti-craving agent.
There are two key factors that operate here.
• First, an effective anti-craving drug for substitution therapy should mimic the agent of abuse at its target protein (receptor or transporter) but it should not do that job too well. Thus, partial direct agonists of a receptor or weak inhibitors of neurotransmitter transport would appear to be the most useful agents. A promising avenue for anti-craving drug development in this niche would be a weaker DA releaser (more like ephedrine than methamphetamine) with a relatively greater effect on DA than on norepinephrine, a moderately rapid onset and a fairly long half-life. Those requirements are met with the transport blockers modafinil and perhaps bupropion.
• Second, the drug’s route of administration needs to allow effective brain concentrations to be reached in a reasonable amount of time (but not too quickly) to avoid further acceleration of the addictive process. Therefore, it will be important to look at the pharmacokinetics of methadone, buprenorphine, nicotine (in its various delivery forms), and modafinil to glean information about the kinetics of these agents.
Reversal Therapy: Substitution therapy has value in that it can stabilize the dependent person at his/her current level of addiction and allow them to function in society. However, substitution therapy often does not allow the person to stop using drugs altogether and, thus, does not affect a “cure”. Ideally, medications would not just reduce craving per se, but also reverse the addictive process, leading to an actual normalization of the person’s brain functions – a true reversal of drug dependence.
The essential feature of the effective anti-craving medications acamprosate and naltrexone (as well as those used off-label, including ondansetron, topiramate, nalmefene, and valproic acid)[48] is that
• They seem to target some portion of the “addictive pathway” that may be common to a variety of addictive drugs and
• They may reverse some of the maladaptive processes associated with the “encoding” of addiction.
This opens an entire avenue of potential treatment options based on a current detailed knowledge of the brain substrates of addiction[7,4] along with knowledge of what drug mechanisms of action are effective in stabilization of brain activity (restoring the balance between excitation and inhibition). As an example, only three drugs are actually approved to reduce ethanol consumption in people with severe alcohol-use disorder (aka alcoholics). These are acamprosate and naltrexone, both acting to reduce craving, and disulfiram, acting to reduce consumption because of the toxic effects that drinking produces in its presence. As discussed in an earlier section, acamprosate and naltrexone have very different mechanisms of action. Acamprosate acts as an NMDA (glutamate) receptor partial co-agonist. Thus, it is an effective modulator of maximal NMDA receptor activity while still providing some degree of tonic stimulation. Naltrexone is a competitive antagonist at opioid receptors, in particular the mu receptor but can modulate DA activity indirectly. Thus, at a molecular level the two drugs couldn’t be more different. However, each agent appears able to reduce the net imbalance in firing within the VTA positive reinforcement pathway towards a more normal value. That may be the key! A summary of the agents discussed thus far is given in Table 3.
Table 3: Anti-Craving Drugs
| TRANSMITTER SYSTEM | THERAPEUTIC DRUG | ADDICTIVE DRUG | MOA – EFFECT ON CRAVING | REFERENCE |
|---|---|---|---|---|
| DOPAMINE [DA] | METHYLPHENIDATE | COCAINE | DAT BLOCKER –NOT HELPFUL | [1] |
| BUPROPION | NICOTINE | DAT/NET BLOCKER – HELPFUL | [2,3] | |
| GAMMA-HYDROXYBUTYRATE | ETHANOL | BLOCKS DA RELEASE – HELPFUL | [4] | |
| NALOXONE | ETHANOL | OPIOID ANTAGONIST – HELPFUL | [4] | |
| MODAFINIL | COCAINE | WEAK DAT BLOCKER – HELPFUL | [5] | |
| SEROTONERGIC [5HT] | 5HT ANTAGONISTS – OSMEMOZOTAN, RITANSERIN, AZASETRON | STIMULANTS | BLOCKS VARIOUS 5HT RECEPTORS ESP 5HT2 – HELPFUL IN RODENTS | [6] |
| ONDANSETRON | ETHANOL | 5HT3 RECEPTOR BLOCKER- VERY HELPFUL IN SUBSET OF ALCOHOLICS | [7] | |
| AMPEROZIDE | ETHANOL | 5HT2 ANTAGONIST – HELPFUL IN RODENTS | [8] | |
| NOREPINEPHRINE [NE] | LOFEXIDINE | COCAINE | ALPHA2 AGONIST – HELPFUL | [9] |
| ACETYLCHOLINE [ACH] | VARENICLINE | NICOTINE | NICOTINIC PARTIAL AGONIST – HELPFUL | [2] |
| GABA | TIAGABINE | ETHANOL | GABA UPTAKE BLOCKER – NOT HELPFUL IN NON-ALCOHOLICS | [10] |
| BACLOFEN | ETHANOL & NICOTINE | GABA-B RECEPTOR DIRECT AGONIST – HELPFUL | [11] | |
| SODIUM CHANNELS | TOPIRAMATE | ETHANOL | BLOCKS VOLTAGE-GATED SODIUM CHANNELS – HELPFUL | [5] |
| LAMOTRIGINE | ETHANOL | BLOCKS VOLTAGE-GATED SODIUM CHANNELS – HELPFUL | [5] | |
| NMDA GLUTAMATE | ACAMPROSATE | ETHANOL, NICOTINE, OPIOIDS | NMDA RECEPTOR PARTIAL CO-AGONIST – HELPFUL | [11,12,5] |
| D-CYCLOSERINE | COCAINE & NICOTINE | NMDA RECEPTOR CO-AGONIST – HELPFUL | [5] | |
| MEMANTINE | NICOTINE & HEROIN | NMDA RECEPTOR NON-COMPETITIVE ANTAGONIST – HELPFUL | [5] | |
| N-ACETYLCYSTEINE | COCAINE | ELEVATES EXTRACELLULAR GLUTAMATE LEVELS – SLIGHT EFFECT | [5] | |
| OPIOID | METHADONE | HEROIN | OPIOID FULL AGONIST – HELPFUL | [13] |
| BUPRENORPHINE | HEROIN | OPIOID PARTIAL AGONIST – HELPFUL | [14] | |
| NALOXONE, NALTREXONE | HEROIN & ETHANOL | OPIOID ANTAGONISTS – HELPFUL | [15,4,3] | |
| CANNABINOID AGENTS | RIMONABANT | NICOTINE | CANNABINOID-1 RECEPTOR INVERSE AGONIST – HELPFUL IN RODENTS REMOVED FROM MARKET FOR PEOPLE | [16] |
| OREXIN | SUVOREXANT | POSSIBLE MULTIPLE DRUGS | OREXIN A/B RECEPTOR ANTAGONIST – NOT YET TESTED | [17] |
- 1. Dursteler, K.M., et al. Clinical Potential Of Methylphenidate In The Treatment Of Cocaine Addiction: A Review Of The Current Evidence (2015) Subst Abuse Rehabil 6: 61-74.
- 2. Islam, N., Rahman, S. Improved Treatment Of Nicotine Addiction And Emerging Pulmonary Drug Delivery. (2012) Drug Discov Ther 6(3): 123-132.
- 3. O'brien, C.P. Anticraving Medications For Relapse Prevention: A Possible New Class Of Psychoactive Medications. (2005) Am J Psychiatry 162(8): 1423-1431.
- 4. Nava, F., et al. Comparing Treatments Of Alcoholism On Craving And Biochemical Measures Of Alcohol Consumptionst. (2006) J Psychoactive Drugs 38(3): 211-217.
- 5. Olive, M.F., et al. Glutamatergic Medications For The Treatment Of Drug And Behavioral Addictions. (2012) Pharmacol Biochem Behav 100(4): 801-810.
- 6. Ago, Y., et al. Neuropsychotoxicity of Abused Drugs: Effects of Serotonin Receptor Ligands on Methamphetamine- and Cocaine-Induced Behavioral Sensitization in Mice. (2008) J Pharmacol Sci 106(1): 15-21.
- 7. Johnson, B.A. Medication Treatment of Different Types of Alcoholism. (2010) Am J Psychiatry 167(6): 630-639.
- 8. Myers, R.D. New Drugs For The Treatment Of Experimental Alcoholism. (1994) Alcohol 11(6): 439-451.
- 9. Sofuoglu, M., Sewell, R.A. Norepinephrine And Stimulant Addiction. (2009) Addict Biol 14(2): 119-129.
- 10. Fehr, C., et al. Tiagabine Does Not Attenuate Alcohol-Induced Activation of the Human Reward System. (2007) Psychopharmacology (Berl) 191(4): 975-983.
- 11. Lin, S.K. Pharmacological Means Of Reducing Human Drug Dependence: A Selective And Narrative Review Of The Clinical Literature. (2014) Br J Clin Pharmacol 77(2): 242-252.
- 12. Jung, Y.C., Namkoong, K. Pharmacotherapy For Alcohol Dependence: Anticraving Medications For Relapse Prevention. (2006) Yonsei Med J 47(2): 167-178.
- 13. Kreek, M.J., et al. Pharmacogenetics And Human Molecular Genetics Of Opiate And Cocaine Addictions And Their Treatments. (2005) Pharmacol Rev 57(1): 1-26.
- 14. Fishman, M.J., Wu, L.T., Woody, G.E. Buprenorphine for Prescription Opioid Addiction in A Patient With Depression and Alcohol Dependence. (2011) Am J Psychiatry 168(7): 675-679.
- 15.










