
Niemann-Pick Disease Type C, A Rare Cause of Pancytopenia: A Case Report
Naima Sghaeir1*, Samia Younes1, Rafik Machraoui1, Samira Younes2, Mohamed Habib Sfar1
Affiliation
- 1Department of Internal Medicine and Endocrinology, Taher Sfar University Hospital, Mahdia, Tunisia
- 2Department of Psychiatry, Taher Sfar University Hospital, Mahdia, Tunisia
Corresponding Author
Naima Sghaeir, Department of Internal Medicine and Endocrinology, Taher Sfar University Hospital, Mahdia, 5100, Tunisia, Tel: 0021673671744; E-mail: naimasghaeir@yahoo.fr
Citation
Sghaeir, N., et al. Niemann-Pick Disease Type C, A Rare Cause of Pancytopenia: A Case Report. (2018) Int J Hematol Ther 4(1): 13- 15.
Copy rights
© 2018 Sghaeir, N. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Niemann-Pick disease type C; Pancytopenia; Poly-lymphadenopathy; Splenomegaly
Abstract
Niemann-Pick disease type C (NPC) is a rare lysosomal storage disease where impaired intracellular lipid trafficking leads to excess storage of cholesterol, sphingomyelin, lycosphingolipids, and sphingosine in tissues. The disease is caused by mutations in one of the two genes, NPC1 or NPC2. Lymphadenopathy and hematological especially pancytopenia in newborns or toddlers with NPC have been anecdotally reported, with limited evidence. The association of this two systemic signs must be differentiated from others blood disorders especially hematological malignancy. We report a case of NPC, in a 26-year-old girl, who presented at the age of 16 a cervical bilateral poly-lymphadenopathy, associated with hematological disorder in which all three major formed elements of the blood (red blood cells, white blood cells and platelets) are decreased in number. The case was essentially reported because of its rare occurrence (pancytopenia and poly-lymphadenopathy which are very rare systemic signs or even exceptional), and because it emphasizes the need for further studies in order to understand how defects in lipid metabolism in NPC can lead to hematological defects especially pancytopenia.
Introduction
Niemann-Pick disease (NPC) is a rare, autosomal recessive and metabolic disease. The prevalence of NPC in early life is probably underestimated, due to high fatality rates and to wide clinical heterogeneity of the disease. The annual rate of NPC is estimated at 1 per 120,000 live births[1]. The incidence of NPC in Tunisia has not been established yet. Approximately 90 – 95% cases of NPC arise from mutations in the NPC1 gene (mapped at 18q11), and 4% result from mutations in the NPC2 gene (mapped at 14q24.3)[2]. The NPC1 protein is in charge of the transport of glycosphingolipids, sphingosine and cholesterol between the cell interior and the cell periphery. This abnormal lipid transport leads to accumulation of lipids in diverse tissues[3]. Therefore NPC has an extremely heterogeneous clinical presentation that ranges from a neonatal rapidly fatal disorder to an adult chronic neurodegenerative disease[2]. These clinical forms may be combined, in rare cases, with hematological defects or poly-lymphadenopathy especially in the infantile form. We report a case of NPC, in a 26-year-old girl, who had pancytopenia and bilateral cervical lymphadenopathy.
Case Report
A 26-year-old woman without a pathological family history, with non consanguineous parents and with no personal history of any other relevant illness, presented at the age of 6, psychomotor deterioration, epilepsy and staturo-ponderal delay. Unfortunately, Seizures could not be controlled with multiple antiepileptic drugs. At the age of 16, she was hospitalized for refractory seizures. Neurological examination found a moderate cerebellar ataxia, dysphagia, deep mental delay, vertical supranuclear ophthalmoparesis, muscular atrophy and areflexia in lower limbs. She had a facial dysmorphia, cervical bilateral poly-lymphadenopathy and splenomegaly. The brain MRI showed cortico-subcortical cerebral and cerebellar atrophy (Figure. 1) with periventricular and subcortical white matter hyperintensity on FLAIR (Figure. 2) and T2 sequences. Electroneuromyographic study demonstrated an axonal peripheral neuropathy. Biological tests detected a pancytopenia (Hb: 9g/dl, L: 2380/mm3 and PLQ: 134.000/mm3) and a low level of cholesterol and LDL-cholesterol (respectively: 2,25 mmol/l, and 0,51 mmol/l). Thoracic and abdominal CT scan showed multiple lymphadenopathies above and below the diaphragm. Lymph node biopsy found lipid surcharge (Figure. 3). The combination of neurological signs (cerebellar syndrome, vertical supranuclear ophthalmoparesis, epilepsy and psychomotor delay), systemic signs (splenomegaly, pancytopenia and lymphadenopathies) and histological findings is very evocative of NPC disease. The patient received symptomatic treatment and anticonvulsant treatment (vigabatrin, phenobarbitol and valproate) with moderate improvement of the epilepsy but not the rest of the signs especially hematological signs.
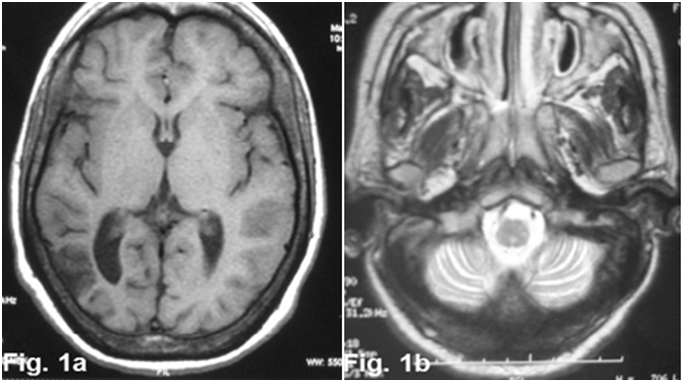
Figure 1: axial cerebral MRI images T1 sequence (1a) and T2 sequence (1b) showed enlargement of cortical sulci and ventricles secondary to cerebral (1a) and cerebellar atrophy (1b).
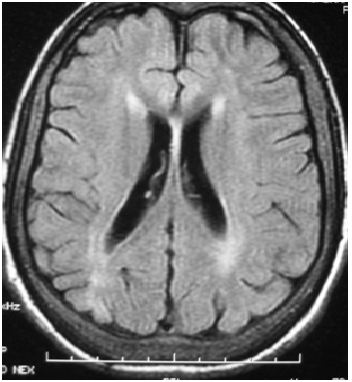
Figure 2: axial FLAIR-weighted cerebral MRI showed periventricular and subcortical white matter hyperintensity with cortical atrophy.
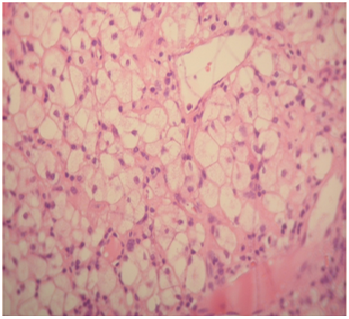
Figure 3: Lymph node biopsy, showed lipid surcharge, in macrophages
Discussion
NPC is caused by mutations in one of the two genes, NPC1 or NPC2, resulting in an accumulation of cholesterol in the endosomal-lysosomal system. Approximately 90 – 95% cases of NPC result from mutations in the NPC1 gene (mapped at 18q11), and 4% arise from mutations in the NPC2 gene (mapped at 14q24.3)[2]. Unfortunately in our case, these mutations were not detected for lack of means. The NPC1 protein is in charge of the transport of glycosphingolipids, sphingosine and cholesterol between the cell interior and the cell periphery. This protein deficiency leads to accumulation of lipids in diverse tissues[3].
It has been shown that visceral symptoms, in particular splenomegaly and prolonged neonatal jaundice, are among the most common symptoms in patients before the age of 4 years, whereas neurological and psychiatric symptoms are predominant in older children and teenagers (4 – 16 years old) and adults (after the age of 16)[2,4]. Our patient’s symptoms began at the age of 6 with neurological signs and got worse at the age of 16 with systemic ones such as splenomegaly, pancytopenia and lymphadenopathies.
The diagnosis of NPC is confirmed by biochemical testing that demonstrates impaired cholesterol esterification and positive filipin staining in cultured fibroblasts[3,5]. Unfortunately, this test is unavailable in our country for luck of means. For our case, the combination of neurological signs (cerebellar syndrome, epilepsy, vertical supranuclear ophthalmoparesis and psychomoteur delay), systemic signs (splenomegaly, pancytopenia and lymphadenopathies) and histological findings is very evocative of NPC disease.
According to Patterson MC[5] the suspicion index should be of use in detecting ‘possible NP-C’ in non specialist centers with limited experience of the disease, once more common diseases have been ruled out. This patient had a score index > 70, indicate a high likelihood of NP-C.
In NPC, accumulation of lipids in tissues could be seen not only in reticuloendothelial and nervous systems, but also in all systems especially lymph nodes. There is a recent study published in 2017, which report the first case of NPC with cervical lymphadenopathy[6], so that our case will be the second case to our knowledge. Based on this case we recommend examination of all lymph nodes if NPC disease is suspected.
The association of poly lymphadenopathy and pancytopenia in NPC must be differentiated from others blood disorders especially hematological malignancy.
Hematological defects such as thrombocytopenia and anemia in newborns or toddlers with NPC have been rarely reported. As far as we know, no study has ever reported a case of Niemann Pick disease with hematological disorder in which all three major formed elements of the blood (red blood cells, white blood cells and platelets) are decreased in number.
Historical or current isolated unexplained splenomegaly, with or without hepatomegaly, is observed in the majority of patients with NPC and it is the strongest visceral indicator of the disease. When combined to other neurological and/or psychiatric symptoms, including ataxia, ophtalmoplegia and schizophrenia-like symptoms, isolated splenomegaly becomes highly suggestive of NPC[1].
Whereas, mild thrombocytopenia is non-specific, as
platelet abnormalities are very common in cases of splenomegaly and have been described in other lysosomal storage diseases.
Bone marrow infiltration with foam cells may cause platelet abnormalities for newborns and these cells can be detected by bone marrow aspiration. Usually, patients with classical foamy cells are the most severely affected ones and they present a large splenomegaly, low platelet counts, bone infiltrates, and the most widespread presentation. It should be noted, however, that patients may rarely present classical foamy cells and splenomegaly at an early age, yet their neurological lesions will remain asymptomatic[1]. However, our patient’s hematological signs are associated with other visceral symptoms, in particular splenomegaly, at the age of 16. Whereas neurological signs have been present since the age of 6.
A study done in 2011, found that C57BL/6J-Npc1/ mice have a shorter lifespan and a very aggressive spleen pathology which correlates with red and white cell abnormalities[4].
In addition, another recent study demonstrated that Zebra fish embryos depleted for NPC1 showed abnormal numbers of thrombocytes and red blood cells, suggesting that the entire myeloid lineage was affected[7,8]. Although, all these findings have yet to be confirmed.
Therapeutically, Miglustat (Zavesca) is a targeted treatment for neurological deterioration in NPC[9] and we hope find a therapeutic alternative for systemic signs.
Conclusion
In Nieamann-pick disease, hematological symptoms are rarely described, whereas their presence plays an important role in the diagnostic orientation and in the prognostic of the disease. The way these defects in lipid metabolism caused by NPC mutations lead to defects in the myeloid lineage remains an open question.
References
1. Mengel, E., Klunemann, H.H., Lourenco, C.M., et al. Niemann-Pick disease type C symptomatology: an expert-based clinical description. (2013) Orphanet J Rare Dis 17: 8-166.
2. Mengel, E., Pineda, M., Hendriksz, C.J., et al. Differences in Niemann-Pick disease Type C symptomatology observed in patients of different ages. (2017) Mol Genet Metab 120(3): 180-189.
3. Vanier, M.T. Niemann-Pick disease type C. (2010) Orphanet J Rare Dis 5:16.
4. Julio, P., Andrés, D.K, Juan, C., et al. Npc1 deficiency in the C57BL/6J genetic back ground enhances Niemann-Pick disease type C spleen pathology. (2011) Biochem Biophys Res Commun 413(3): 400-406.
5. Patterson, M.C., Hendriksz, C.J., Walterfang, M., et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. (2012) Mol Genet Metab 106(3): 330-344.
6. Inci, A., Okur, I., Esendagli, G., et al. Patient with Niemann-Pick Type C Presenting With a Jaw Mass Characterized With Lymph Node Involvement by Niemann-Pick Cells. (2017) J Pediatr Hematol Oncol
7. Louwette, S., Régal, L., Wittevrongel, C., et al. NPC1 defect results in abnormal platelet formation and function: studies in Niemann-Pick disease type C1 patients and zebrafish 2013. (2013) Hum Mol Genet 22(1): 61-73.
8. Louwette, S., Régal, L., Wittevrongel, C., et al. Niemann-Pick disease and platelet dysfunction 2012. (2012) Dis Model Mech 5(6): 808.
9. Bowman, E.A., Velakoulis, D., Desmond, W.M. Longitudinal changes in white matter fractional anisotropy in adult-onset Niemann-Pick disease type C patients treated this Miglustat. (2017) JIMD Resp 1-5.










