
Pathway-Based Approaches for Analysis of Genome-Wide Association Studies -A Case Report for Metastatic Small Cell Lung Cancer
JingLu1* ShuQun Cheng2*, Biaoru Li3#
Affiliation
- 1OBGYN, Monterey Park, CA 91754, USA
- 2Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, Shanghai, 200438, P.R. China
- 3Georgia Cancer Center, MCG, Augusta, GA, USA 30912
Corresponding Author
Biaoru Li, Department of Pediatrics, Section of Hematology/Oncology, Georgia Cancer Center, MCG, Augusta, GA, USA 30912; E-mail: BLI@gru.edu
Citation
Li, B., et al. Pathway-based Approaches for Genome-Wide Association Studies - A Case Report for Personalized Therapy to Metastatic Small Cell Lung Cancer. (2017) Int J Hematol Ther 3(2): 1- 8.
Copy rights
© 2017 Li, B. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License
Keywords
SNP signature; Quantitative network; Drug discovery; Betweenness centrality (BC); Degree centrality (DC)
Abstract
Lung cancer is a common malignant tumor including Small Cell Lung Cancer (SCLC) with 10 - 15 % and Non-Small Cell Lung Cancer (NSCLC) with 70 - 80%. Currently, there are several approaches to be used to treat lung cancer including surgery, chemotherapy, radiation therapy and molecular therapy/immunotherapy, Metastatic SCLC, metastatic mixed type of lung cancer and unclassified lung cancer are still difficult to be cured because this kind of lung cancer is easy to be widely disseminated. For example, if a patient has several metastatic SCLC, the patient will reveal poor outcome. In order to resolve the poor prognosis with recurrent and metastatic SCLC, here we reported a pathway-based approaches for analysis of Genome-Wide Association Studies (GWAS) to screen drugs, hence we used the drugs to treat a patient suffering from SCLC with multiple metastases. In the beginning, we harvested a pair of SCLC cells and normal cells from FFPE samples under laser capture microscopy to achieve the tumor cell DNA for SNP profile. After uncovering SCLC SNP profile, genes related to SNP signature was used to map quantitative network to uncover targeting drugs. Synchronously, the targeting SNPs genes were further confirmed by TaqMan PCR and Sanger’s sequencing. Furthermore, selected drugs were also validated for the SCLC cells by computational modeling. Finally, the drugs approved by FDA were administered for the patient to the personalized therapy. All in all, after the drugs were discovered by GWAS profiles through quantitative network, we successfully achieve a good response for the patient who has suffered from SCLC with multiple metastases
Introduction
Personalized medicine is a new generation model to be directly tailored for physicians to care individual patient relying on personal genomic profiles[1]. It is often called as “the right treatment for the right person at the right time.” A satisfactory personalized therapy should have a rational genomic modeling to achieve sensitive drugs for clinical applications[2]. Following Research and Development (R&D) of genomic modeling and analysis, clinical sampling along with system modeling has been increasingly reported for personalized therapy[3]. Here we introduce a pathway-based approach for analysis of Genome-Wide Association Studies (GWAS) to screen drugs, and thus we report a case by using the model including clinical sampling by Laser Capture Microscopy (LCM) to harvest Small Cell Lung Cancer (SCLC) and normal cells from FFPE tissues (Formalin-Fixed Paraffin-Embedded tissue), to analyze SNP profiles and then to discover SNP signature related gene by system modeling as well as to uncover sensitive drugs from drug-bank for the patient suffering from SCLC metastasis.
Clinical sampling for genomic analysis of tumor cells consist of a pair of surgical tumor tissue vs normal tissue by in vivo harvest, or from a pair of tumor cells vs normal cells in situ harvest obtained from Laser Capture Microscopy (LCM) or from a pair of cells by ex vivo culture from clinical specimens[4]. Here a pair of SCLC cells vs normal control cells obtained from LCM was used to GWAS and SNP analysis for the SCLC patient. In addition, system modeling concerning SNP signature has the ability to provide some important information required for therapeutic targeting identification and drug discovery[5]. After SNP signature database were combined with quantitative bioinformatics analysis, SNP related genes can provide us to identify therapeutic targeting genes and to discover drugs for patients with drugs sensitivity for the tumor diseases[6]. Here, in order to introduce a case report using pathway-based approaches of Genome-Wide Association Studies (GWAS) to screen drugs for a patient suffering from SCLC with multiple metastases, we present a mining process including harvesting a pair of SCLC cells and normal control cells, performing SNP microarray, mining significant or functional SNP for signature SNP, finally uncovering sensitive drug for the patient application. Following the four steps, that is, clinical sampling, SNP performance and mining SNP profile, discovering SNP signature related genes, uncovering sensitive drugs, finally, a list of sensitive drugs targeting SCLC will be used to personalized chemotherapy for the patient.
Clinical Specimens and Methods
Patient and specimen
The patient was given diagnoses according to clinical criteria. Informed consent of the patient was obtained before tumor tissue sampling. SCLC was diagnosed and classified according to cell type by conventional pathology. After SCLC tissues embedded on slides about 10- μm section was stained with light May-Grunwald-Giemsa staining method(MMG), laser capture microscope (Zeiss Palm Microbeam IV Laser Capture Microscope) identify regions rich in tumor cells and normal tissues as our previous report[7]. The tumor-rich regions and normal cells were isolated by LCM to harvest the specimen DNA.
Genomic DNA and QC Analysis
Because DNA on FFPE slides had been discovered degradation, DNA were amplified and rescued by two processes. Whole-Genome Amplification (WGA) was performed by using the Genome Plex Single Cell Whole Genome Amplification kit (Sigma-Aldrich) according to the manufacturer’s protocol[8]. Briefly, after degraded DNA was harvested in tubes, the sample was complemented to 9 μL nuclease-free water and 1 μL single cell lysis with proteinase K mix. The subsequent incubation for cell lysis (50 °C for 1 h) as described by the manufacturer. WGA products were purified with the GenElute PCR Clean-Up kit (Sigma-Aldrich). After DNA specimens from SCLC and normal control cells were amplified, we also performed DNA rescuing with FFPER estore Protocol Kit (Illumina Inc Santa Clara, CA) according to the manufacturer’s protocol[9]. The amplified and rescued DNA was quantified by Nano Drop 2000 and its amplicon from GAPDH with PCR primers design for 100 bp, 500 bp and 1 kb amplification was demonstrated by PCR with DNA gel running and quantified by DNA UV quantification for rescuing assay.
SNP Microarray Performance
Each 800 ng of DNAs was sent to Genomic core in Kansas University Medical Center to perform SNP 6.0 microarray (Affymetrix SNP 6.0 with 906,600 SNPs) for the experiments[10]. Affymetrix SNP 6.0 was processed using the Genome Alteration Print method to obtain absolute SNP and copy number profiles. Affymetrix Genotyping Console 3.0.2 were used to transfer the signal intensities into CEL files and determine signal intensities. The samples passed the Affymetrix QC threshold as QC Call Rate > 87 % and genotype calls of CEL files were made using Birdseed v2 (Affymetrix GTC software).
Bioinformatics analysis
We first uncovered significant SNP based on the genotype calls from the paired tumor cells and normal cell samples by which we excluded any SNP obtained from normal control cells, leaving tumor SNPs for further analyses. Moreover, because genomic DNA amplification and rescuing were performed and because personalized therapy more focus on SNP-based genotype change, we excluded any LOH and CNV from all tumor cells so that only SNP change from tumor cells was subject to further study SNP-based therapeutic targeting. Important and functional SNP was analyzed by SNP related key genes by SNP annotation software. In order to study SNP targeting therapy, we used two ways to mine SNP related targeting: pathway-based SNP targeting and SNP-direct-drug targeting.
In pathway-based SNP targeting analyses, PINBPA software plugged in Cytoscape 3.3 platform has been implemented for the analysis[11]. All genes related functional SNPs were sorted by their genomic coordinates and defines signature SNP by the platform with threshold cutoff (p-value < 0.05). After harvesting signature SNP related genes, a python platform, which we have published[12], discover the signature SNP related genes and drugs. Our quantitative analysis and network topology focused on Betweenness Centrality (BC) and Degree Centrality (DC) as our previous report[13].
Validation of Signature SNP with their Drugs for Clinical Application
SNP-direct-drug targeting: After signature SNPs profiles were uncovered, GeneGo software and Genebank were used to search drugs[14], several sensitive drugs related signature SNP for SCLC was directly discovered to use for targeting SCLC.
Pathway-based SNP targeting: Drugs related with therapeutic identification SNP and genes was identified by Python-based platform which we have published for network topology with computational analysis for specifically targeting the SCLC[15].
Validation of signature SNP: TaqMan genotype technique was used to confirm SNPs and Sangers sequencing was used for SNP detection related with the assay of specificity and sensitivity. Both techniques were performed according to the manufacturer’s and equipment protocol (Thermo Fisher Scientific)[16].
Results
Patient information
The patient is 58 years old male. In December, 2015, he had cough with blood streak in sputum. Multiple masses were found with increase of FDG uptake in the inferior lobe of left lung by PET/CT. In addition, multiple bone destructions were seen in bilateral ribs, C2, T1, T9-10, L3-4 vertebral bodies, bilateral iliac bones, bilateral pubic bones, right acetabular bone, left caput femoral bone including FDG uptake obviously increased in the cervical, thoracic and lumbar vertebra. Liquid-based cystoscopy from biopsy reported that poorly differentiated carcinoma, small cell carcinoma in mediastinum lymph nodes.
Results of genomic DNA performance and SNP microarray
In order to mine SNP profiles from patient specimens, we used LCM technique to harvest a pair of cells under MMG staining for 1 min. as Figure-1A-I. Total 28 SCLC clusters with 93 % tumor cells as Figure-1A-II and 15 normal control cells cluster were harvested as Figure-1A-III. Because DNA on FFPE slides had been discovered degradation as Figure-1B-I. After WGA and DNA recuing process as Figure-1B-II, amplified and rescued DNA demonstrated recovery rate 90 - 92 % in 100 bp amplicon, 78 - 79 % in 500 b amplicon and 56 - 58 % in 1 kb, respectively, as Figure-C. Each 800 ng of DNAs (T-1 and T-2) was performed for SNP 6.0 microarray (Affymetrix SNP 6.0 with 906, 600 SNPs) for the experiments. Affymetrix Genotyping Console 3.0.2 were used to mine SNP and CNV with QC (QC Call Rate > 87 % ) under Birdseed v2 (Affymetrix GTC software) as Table-1.
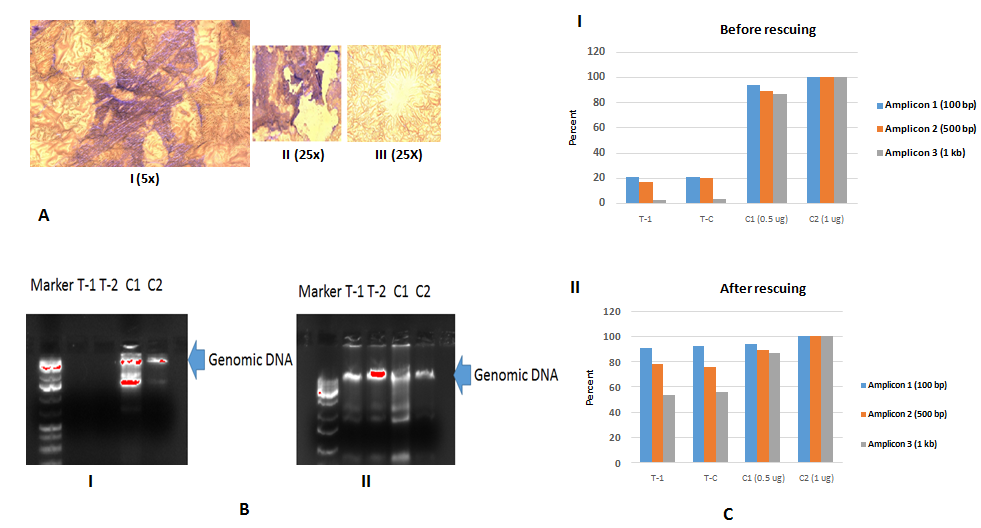
Figure 1: A-I Giemsa stain with 1 minute staining is under 5x magnificence microscope before LCM; A-II is SCLC cell clusters and A-III is normal control cell cluster which were harvested by LCM.B-I is DNA gel: T1 and T2 demonstrate DNA degradation to compare C1 (LCM pick up Hela cell as fixed Hela cells) and C2 (culture Hela cells as living cells); B-II is DNA gel. T1 and T2 demonstrate DNA are rescued as to compare C1 (LCM pick up Hela cell) and C2 (culture Hela cells); C shows that DNA amplicon from GAPDH genes which are designed length with 100 bp, 500 bp and 1000 bp; Top picture in C-I demonstrate DNA degradation at 100 bp, 500 bp and 1 kb and bottom picture DNA in C-II are rescued at 100 bp, 500 bp and 1 kb to compare C1 (LCM pick up Hela cell) and C2 (culture Hela cells).
Table 1: QC results for GWAS.
| File | Bounds | Contrast QC | QC Call Rate | CHP/CEL | |
|---|---|---|---|---|---|
| 1 | T1_061416_(GenomeWideSNP_6).CEL | In | 1.82 | 93.75 | 1 |
| 2 | T2_061416_(GenomeWideSNP_6).CEL | In | 1.71 | 93.85 | 1 |
Results of GWAS
After both SCLC SNP (called as T-1) and normal control cells SNP (called as T-2) were performed by Affymetrix SNP 6.0 chip, each with total 906,600 SNPs, 1425 SNPs (only from tumor cells) were discovered as Supplement_Table-1, and thus the results are demonstrated as Figure-2. We also mined CNV (copy number variances) from all tumor cells and normal control cells as Figure-3A and Figure-3B in which green colors indicating gaining variances and red color meaning lose variances. Total 1104 CNV all tumor cells were discovered as Figure-3C. Because current personalized therapy was designed by pathway-based approaches from GWAS, SNP-based genotype change from tumor cells was used to analysis, and therefore 924 gene-related to SNP (called as functional SNPs) were uncovered as Supplement Table-2. In pathway-based approaches from the signature SNP, their genes analysis was implemented by PINBPA software plugged into Cytoscape 3.3. All genes related functional SNPs were sorted by their genomic coordinates and defines association blocks at user-defined threshold (p-value < 0.05).
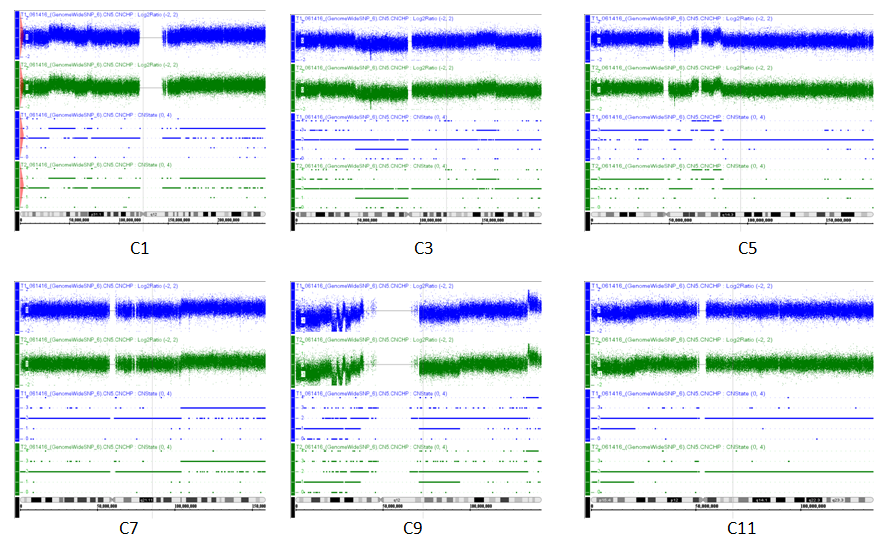
Figure 2: T1 (blue) and T2 (green) results from SNP and CNV microarray performance and analysis indicating chromosomes are differences between tumor sources and normal control. Because chromosomes are largely different at chromosomes1, 3, 5, 7, 9, 11, fig-2 show C1, C3, C5, C7, C9, C11 changes at CNV and SNP.
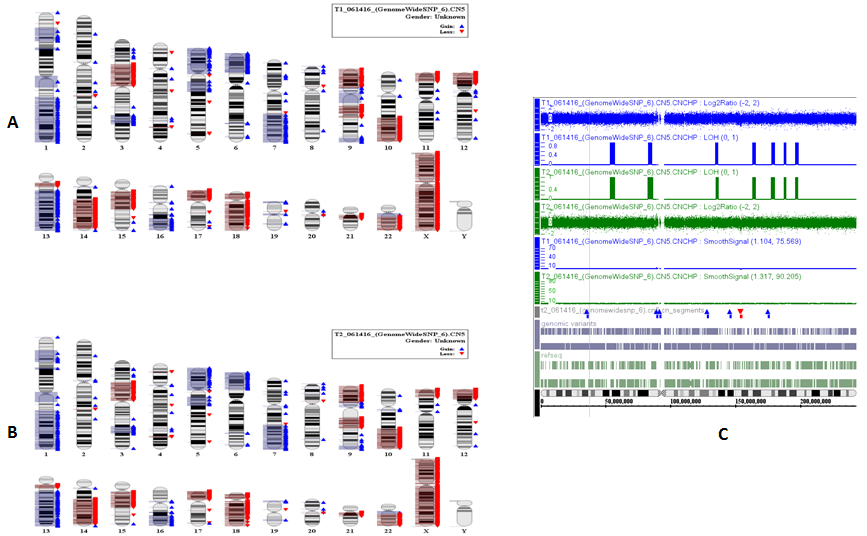
Figure 3: A demonstrates CNV from T-1 and B means CNV from T-2.
Table 2: SNP directly Targeting Genes and Drugs.
| Associated Gene | dbSNP RS ID | Chromosome | Targeting drugs | Targeting molecular therapy |
|---|---|---|---|---|
| ALK | rs12052994 | 2 | carboplatin | crizotinib, ceritinib |
| KRAS | rs10505978 | 12 | carboplatin, paclitaxel, pemetrexed, docetaxel | erlotinib |
| MEGF10/11 | rs9327438/rs10518678 | 5_15 | erlotinib, gefitinib, and afatinib | |
| TP53I3 | rs2303287 | 2 | docetaxel |
Drug discovery
After harvesting signature SNP related genes, SNP-direct-drug targeting were uncovered by GeneGo software and Genebank. In SNP-direct-drug targeting, we found several drugs and molecular therapy for KLF, KRAS and EGF and TP53I3 related SNP such as Carboplatin at Table-2.
In order to study a practicable platform, Betweenness Centrality (BC) indicates effect and Degree Centrality (DC) means both effect and side-effect under a python-based platform as our previous study. Accordingly, pathway-based SNP targeting were used to analyze sensitive and specific drugs, and therefore a list of sensitive drugs was discovered by the network topology as Table-3. As shown in Table-3, cisplatin, irinotecan and etoposide are top sensitive to targeting SCLC cells from the patient. The results of pathway-based GWAS analysis including targeting genes, drugs and SCLC cells was performed as Figure-4A1 and Figure-4A2 and mined as Figure-4B with computational mimic analysis under Python scripts to confirm specifically targeting to the patient SCLC cells.
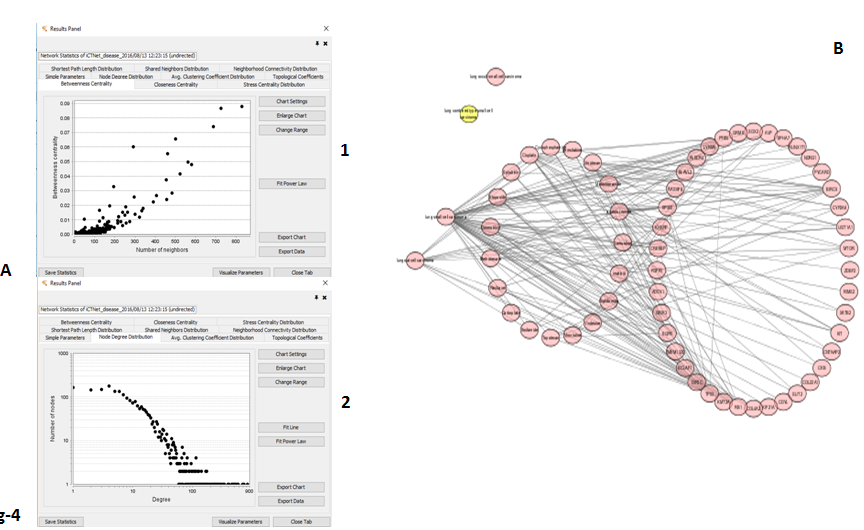
Figure 4: A-1 is Betweenness scatter plot and A-2 is Degree scatter plot; B is network results from gene related SNP signatures in SCLC cells including genes, drugs and SCLC cells. Pink color is network indicating the genes-drugs related to SCLC.
Table 3: Pathway-based approaches for GWAS.
| Name | Betweenness Centrality | Degree Centrality |
|---|---|---|
| Cisplatin | 0.06046899 | 18 |
| irinotecan | 0.01758476 | 10 |
| Etoposide | 0.01395787 | 10 |
| Ifosfamide | 0.00967844 | 4 |
| imatinib | 0.00753371 | 6 |
| Methotrexate | 0.00741667 | 7 |
| Topotecan | 0.00695399 | 5 |
| Doxorubicin | 0.00575564 | 10 |
| Vincristine | 0.00438819 | 4 |
| Paclitaxel | 0.00417765 | 7 |
| Cyclophosphamide | 0.00361606 | 4 |
| gemcitabine | 0.00355958 | 6 |
| Vindesine | 0.00328222 | 2 |
| Epirubicin | 0.00154738 | 4 |
| Thalidomide | 0 | 3 |
| Carboplatin | 0 | 2 |
| Metoclopramide | 0 | 2 |
| Lomustine | 0 | 1 |
Drugs verification and clinical results
After mining therapeutic target and discovering drugs, finally the both groups of SNP signature were confirmed by TaqMan PCR, and thus SNP sensitivity (93 % ) and specificity (68.3 % ) were supported by Sanger sequencing.
Target drugs related to SNP signature genes, which have been approved by FDA, were used to the patient. According to drug list from pathway-based SNP targeting, in which cisplatin and etoposide combination therapy have been approved by FDA, 6 courses cisplatin and etoposide were administered to the patient. After 6 courses of cisplatin and etoposide were administered to the patient as Table-4, tumor masses in left lung was decreased from 3 x 4 cm into 0.8 x 1.1 cm as in Figure 5A1 and Figure 5A2 from X-ray. The metastasis in left lung, thoracic vertebra and lumber vertebra have partial responses after the 3 month’s personalized therapy with observation in the following 3 months as Figure 5B and 5C from CT. The patient side-effects were demonstrated as Table 5.
Table 4: Drug Administer.
| Case | Drugs | FDA approval | Dosage | Application methods |
|---|---|---|---|---|
| YZ | VP-16 | Approval | 220 mg d1-3 x6 | intravenous |
| DDP | Approval | 56 mg d1-3 x6 | intravenous |
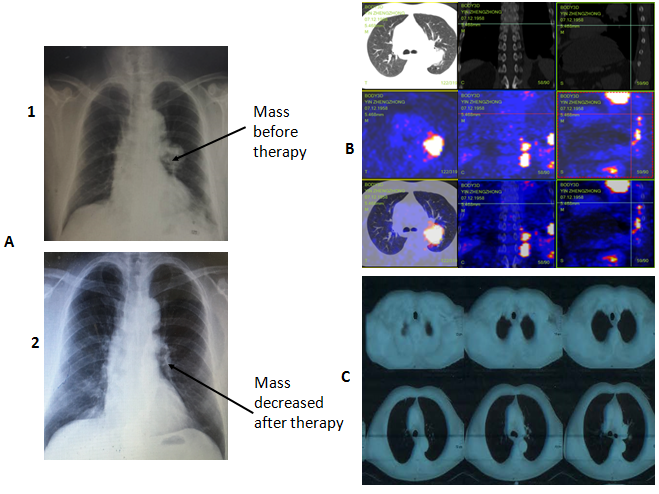
Figure 5: A-1demonstrates X-ray results before treatment with a large mass in left lungs and A-2 demonstrates X-ray results after treatment with a mass decrease in left lungs; B shows PET/CT results indicating multiple metastases in left lung, thoracic vertebra and lumber vertebra; C demonstrates partial responses in CT picture after three months of personalized chemotherapy.
Table 5: Drug side-effect.
| Side-effect | Course-1 | Course-2 | Course-3 | Course-4 | Course-5 | Course-6 |
|---|---|---|---|---|---|---|
| Grade of Adverse Events | 3 | 2 | 2 | 2 | 2 | 1 |
| Leukopenia | Yes | Yes | Yes | Yes | Yes | Yes |
| Nausea and vomiting | Yes | Yes | Yes | Yes | Yes | Yes |
| weight gain | Yes | Yes | Yes | Yes | Yes | Yes |
| Alopecia | Yes | Yes | Yes | Yes | Yes | Yes |
| Cardiac function | No | No | No | No | No | No |
| Edema | No | No | No | No | No | No |
| Thrombocytopenia | No | No | No | No | No | No |
| Shock, sepsis | No | No | No | No | No | No |
| Systemic infection | No | No | No | No | No | No |
Discussion
Major advances in human genome with their techniques provide our ability to identify SNPs related to the prevalence of genetic, tumor and some rare diseases[17]. Three ways can detect SNPs based on a given laboratory with their laboratory conditions. RNA-Seq can discover both global SNPs and transcriptome profiles, DNA-Seq can achieve global and unknown SNP data, and SNP microarray should harvest global SNPs due to the priori information[18]. After SNPs are detected by global methods, GWAS pairing disease-control samples with a statistical analysis can determine whether the alleles at the marker can predict the phenotype alteration. If GWAS reaches statistical significance after multiple testing, the variant is considered to be associated with the diseases. Now SNPs has been successfully conducted by GWAS to predict disease risks[19]. In addition, following risks prediction to GWAS to identify SNP related with a given disease, functional SNPs techniques are further studied for causal SNP discovery. GWAS related to functional SNPs play an important role in pathogenesis, prediction and precaution of diseases[20]. As reported in 2014, almost 2,000 publications have reported associations of more than 13,000 Single Nucleotide Polymorphisms (SNPs) with close to 200 phenotypes in the GWAS Catalog[21]. The successful record of this genomic mapping strategy includes the identification of dozens or even hundreds of susceptibility alleles in common diseases such as multiple sclerosis, type 1 and type 2 diabetes, lymphoma, leukemia, and metabolic disorders[22].
Despite GWAS has been extensively used to identify susceptibility in common diseases, this is a necessary to develop GWAS-link, global and individual, of phenotype markers. These approaches called as pathway analysis aim at identifying functional relationships among GWAS associated proteins and diseases. Fortunately, after these data have been quickly integrated, such as SNP-protein, SNP-phenotype, protein-protein and gene/ protein-drugs, a network has emerged in SNPs profiles called as “SNP-gene-protein-phenotype-drugs” combined by system biology[23]. Now, personalized medicine related with pharmacogenomics information can predict drugs response from individual Single Nucleotide Polymorphisms (SNP) and Genome-Wide Association Studies (GWAS)[24]. The interdisciplinary method can construct systems to guide a suggested treatment for personalized medicine. Here, we first set up “Pathway-Based Approaches from Genome-wide Association Studies” based on our previous study, or, higher BC is related with higher effect whereas higher both BC and CD are related higher effect and higher side-effect. On the other hands, higher BC are related with higher effect whereas only lower CD is related with lower toxicity[25].
The patient has multiple metastasis from SCLC so that it is good candidate for personalized therapy. In order to mine SNP profiles specific from SCLC cells, we applied for LCM sampling to harvest SCLC called to compare normal control cells. After we mined SNPs from SCLC, like most GWAS analysis, SNPs from tumor cells (1425 significant SNPs from tumor cells) was subject to further study pathway-based approaches from GWAS data. Although Affymetrix SNP 6.0 chip have total 906,600 SNPs and CNV on the chip, current personalized therapy was designed for SNP-based pathway approaches from GWAS. In pathway-based approaches from GWAS, the SNP related genes were used to discover the signature SNP related genes and drugs. According to mining therapeutic targeting and drugs, clinically, target drugs approved by FDA were selected to the patient treatment. Because cisplatin and etoposide combination therapy have been approved by FDA, six courses cisplatin and etoposide were administered to the patient. After six courses of cisplatin and etoposide were administered for the patient, tumor masses in left lung were decreased and metastasis in left lung, thoracic vertebra and lumber vertebra have partial responses with observation in the following 3 months. This case report demonstrated that a pathway-based approaches for GWAS analysis can successfully achieve a good response for clinical patient.
Author’s Contributions
BL conceived, designed and guided the work and edited the manuscript. JL and SJC involved process with supported clinical work.
Acknowledgement
The inclusion of trade names or commercial products in this article was solely for the purpose of providing specific information and does not imply recommendation for their products.
References
- 1. Schilsky, R.L. Opinion: Personalized medicine in oncology: the future is now. (2010) Nature Reviews Drug Discovery 9: 363-366.
Pubmed || Crossref || Others - 2. Hudson, T.J. Genome variation and personalized cancer medicine. (2013) J Intern Med 274(5): 440-450.
Pubmed || Crossref || Others - 3. Li, B., Senzer, N., Rao, D.D. Bioinformatics Approach to Individual Cancer Target Identification, 11th Annual Meeting of the American Society of Gene Therapy (2008) C8: 451004.
Pubmed || Crossref || Others - 4. Li, B. Clinical Genomic Analysis and Diagnosis-Genomic Analysis Ex Vivo, in Vitro and in Silico. (2012) Clinical Medicine and Diagnostics 2(4): 37-44.
Pubmed || Crossref || Others - 5. Yoshida, T., Ono, H., Kuchiba, A., et al. Genome-wide germline analyses on cancer susceptibility and GeMDBJ database: Gastric cancer as an example. (2010) Cancer Sci 101(7): 1582-1589.
Pubmed || Crossref || Others - 6. Hudson, T.J. Genome variation and personalized cancer medicine. (2013) J Intern Med 274(5): 440-450.
Pubmed || Crossref || Others - 7. Emmert-Buck, M.R., Bonner, R.F., Smith, P.D. Laser capture microdissection. (1996) Science 274(5289): 998-1001.
Pubmed || Crossref || Others - 8. Jochen, B., Geigl, J.B., Speicher, M.R. Single-cell isolation from cell suspensions and whole genome amplification from single cells to provide templates for CGH analysis. (2007) Nat Protoc 2(00312): 3173-3184.
Pubmed || Crossref || Others - 9. Chung, J., Son, D.S., Jeon, H.J., et al. The minimal amount of starting DNA for Agilent's hybrid capture-based targeted massively parallel sequencing. (2016) Sci Rep 6: 26732.
Pubmed || Crossref || Others - 10. Demir, E., Hasdemir, C., Ak, H., et al. Genome-Wide Association Study of Copy Number Variations in Patients with Familial Neurocardiogenic Syncope. (2016) Biochem Genet 54(4): 487-494.
Pubmed || Crossref || Others - 11. Wang, L., Matsushita, T., Madireddy, L., et al. PINBPA: cytoscape app for network analysis of GWAS data. (2015) Bioinformatics 31(2): 262-264.
Pubmed || Crossref || Others - 12. Zheng J., Zhang, D., Przytycki, P.F., et al. SimBoolNet--a Cytoscape plugin for dynamic simulation of signaling networks. (2010) Bioinformatics 26(1): 141-142.
Pubmed || Crossref || Others - 13. Narayanan, T., Gersten, M., Subramaniam, S., et al. Modularity detection in protein-protein interaction networks. (2011) BMC Res Notes 4: 569.
Pubmed || Crossref || Others - 14. Chen, K., Wu, D., Bai, Y., et al. Fuzzy clustering analysis of osteosarcoma related genes. (2014) Pathol Oncol Res (3): 535-539.
Pubmed || Crossref || Others - 15. Hu, H, Shen Li, Zhang, X., et al. A Therapeutic Targeting Identification from Microarray Data and Quantitative Network Analysis. (2015) The Open Access Journal of Science and Technology 3: 101-104.
Pubmed || Crossref || Others - 16. Burns, C.C., Kilpatrick, D.R., Iber, J.C., et al. Molecular Properties of Poliovirus Isolates: Nucleotide Sequence Analysis, Typing by PCR and Real-Time RT-PCR. (2016) Methods Mol Biol 1387: 177-212.
Pubmed || Crossref || Others - 17. Rodríguez-Antona, C., Taron, M. Pharmacogenomic biomarkers for personalized cancer treatment. (2015) J Intern Med 277(2): 201-217.
Pubmed || Crossref || Others - 18. McBean, R.S., Hyland, C.A., Flower, R.L. Approaches to determination of a full profile of blood group genotypes: single nucleotide variant mapping and massively parallel sequencing. (2014) Comput Struct Biotechnol J 11(19): 147-151.
Pubmed || Crossref || Others - 19. Hollegaard, M.V., Grauholm, J., Nielsen, R. Archived neonatal dried blood spot samples can be used for accurate whole genome and exome-targeted next-generation sequencing. (2013) Mol Genet Metab 110(1-2): 65-72.
Pubmed || Crossref || Others - 20. Kim, Y.S., Kim, Y., Choi, J.W., et al. Genetic variants and risk of prostate cancer using pathway analysis of a genome-wide association study. (2016) Neoplasma 63(4): 629-634.
Pubmed || Crossref || Others - 21. Welter, D., MacArthur, J., Morales, J., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. (2014) Nucleic Acids Res 42(Database issue): D1001-6.
Pubmed || Crossref || Others - 22. Smushkin, G., Vella, A. Genetics of Type 2 Diabetes. (2010) Curr Opin Clin Nutr Metab Care 13(4): 471–477.
Pubmed || Crossref || Others - 23. Jain, P., Vig, S., Datta, M., et al. Systems Biology Approach Reveals Genome to Phenome Correlation in Type 2 Diabetes. (2013) PLoS One 8(1): e53522.
Pubmed || Crossref || Others - 24. Saglio, G., Morotti, A., Mattioli, G. Rational approaches to the design of therapeutics targeting molecular markers: the case of chronic myelogenous leukemia. (2004) Ann N Y Acad Sci 1028(1): 423-431.
Pubmed || Crossref || Others - 25. Peng, Q., Schork, N.J. Utility of Network Integrity Methods in Therapeutic Target Identification. (2014) Front Genet 5: 12.
Pubmed || Crossref || Others










