
Potential Contributions of the Tobacco Nicotine-Derived Nitrosamine Ketone to White Matter Molecular Pathology in Fetal Alcohol Spectrum Disorder
Ming Tong1,2, Tomas Andreani1, Alexander Krotow3, Fusun Gundogan2,4, and Suzanne M. de la Monte1,2,3,5*
Affiliation
- 1Department of Medicine, Division of Gastroenterology, and the Liver Research Center Rhode Island Hospital, Providence, RI
- 2Warren Alpert Medical School of Brown University, Providence, RI
- 3Pathobiology Graduate Program, Brown University, Providence, RI
- 4Department of Pathology, Women and Infants Hospital of Rhode Island, Providence, RI
- 5Departments of Pathology and Neurology, and the Division of Neuropathology, Rhode Island Hospital, Providence, RI Supported by AA-11431 and AA-12908 from the National Institute of Alcohol Abuse and Alcoholism of the National Institutes of Health
Corresponding Author
Dr. Suzanne M. de la Monte, MD, MPH, Pierre Galletti Research Building, Rhode Island Hospital, 55 Claverick Street, Room 419, Providence, RI 02903, Tel: 401-444-7364; Fax: 401-444-2939; E-mail: Suzanne_DeLaMonte_MD@Brown.edu
Citation
de la Monte, S.M., et al. Potential Contributions of the Tobacco Nicotine-Derived Nitrosamine Ketone to White Matter Molecular Pathology in Fetal Alcohol Spectrum Disorder (2016) J Neurol Brain Dis 3(2): 1- 12.
Copy rights
© 2016 de la Monte, S.M. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Tobacco; Nitrosamines; Alcoholic brain disease; Adducts; PCR array; Myelin; Neurodegeneration
Abstract
Background: Fetal alcohol spectrum disorder (FASD) is associated with long-term deficits in cognitive and motor functions. Previous studies linked neurodevelopmental abnormalities to increased oxidative stress and white matter hypotrophy. However, similar effects occur with low-dose nitrosamine exposures, alcohol abuse correlates with cigarette smoking, and tobacco smoke contains tobacco-specific nitrosamines, including NNK.
Hypothesis: Tobacco smoke exposure is a co-factor in FASD.
Design: Long Evans rat pups were i.p. administered ethanol (2 g/kg) on postnatal days (P) 2, 4, 6 and/or NNK (2 mg/kg) on P3, P5, and P7 to simulate third trimester human exposures. Oligodendroglial myelin-associated, neuroglial, and relevant transcription factor mRNA transcripts were measured using targeted PCR arrays.
Results: Ethanol and NNK differentially altered the expression of immature and mature oligodendroglial, neuronal and astrocytic structural and plasticity-associated, and various transcription factor genes. NNK’s effects were broader and more pronounced than ethanol’s, and additive or synergistic effects of dual exposures impacted expression of all four categories of genes investigated.
Conclusion: Developmental exposures to alcohol and NNK (via tobacco smoke) contribute to sustained abnormalities in brain white matter structure and function via distinct but overlapping alterations in the expression of genes that regulate oligodendrocyte survival, maturation and function, neuroglial structural integrity, and synaptic plasticity. The results support the hypothesis that smoking may contribute to brain abnormalities associated with FASD.
Introduction
Heavy chronic or binge alcohol exposures during development lead to sustained deficits in cognitive and motor functions. The characteristic constellation of craniofacial abnormalities together with brain structural pathology is referred to as fetal alcohol spectrum disorder (FASD)[1,2]. The cerebellum, temporal lobe, and white matter are major targets of ethanol’s teratogenic effects, and while there is good evidence that ethanol-induced pathology in the cerebellum and temporal lobe is linked to impaired insulin and insulin-like growth factor type 1 (IGF-1) signaling through Akt-mediated growth, cell survival, metabolic, and neuronal plasticity networks, the nature and pathogenesis of white matter hypotrophy are poorly understood. However, since growth factor, including IGF-1 signaling through phosphatidylinositol-3-kinase (PI3K) and Akt promotes oligodendrocyte survival and function, including myelin synthesis, maintenance, and homeostasis[3-5], impairments in insulin/IGF signaling that occur with alcohol exposure could potentially underlie the white matter abnormalities in FASD. Since the white matter hypotrophy may be due to reduced myelin synthesis, maturation and, maintenance or primary loss of axons, evaluating the state of oligodendrocyte function could aid our understanding of its molecular pathogenesis.
Oligodendroglia produces central nervous system (CNS) myelin. Myelin has an abundant lipid composition (70 - 85%) with lower proportions of proteins (15 - 30%) and water compared with other cellular membranes. High lipid content ensures optimum nerve cell conduction. Oligodendrocytes develop from precursor cells (OPC 1-3) that differentially express Cnp, Ng, PDGFRα, Olig2, Dix2, and Nkx. OPCs differentiate into immature oligodendroglia that express Cnp, Olig1, and low levels of Olig2, which mature and express Cnp, Olig1, low Olig2, and RTN4[6-8]. Finally, mature myelinating oligodendroglia further express distinct integral membrane proteins including, myelin basic protein (MBP), myelin associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP)[3,4,9]. Oligodendroglial differentiation is regulated by Wnt/β-catenin (canonical pathway) via the Tcf4 transcription factor[10], Notch1-HES5, and insulin/IGF signaling[11-19].
Chronic ethanol exposure delays oligodendroglial maturation, expression of MBP[20] and MAG[21], and de novo myelin synthesis[22]. White matter integrity is crucial for CNS function since reduced brain myelination during development results in cognitive impairment[23]. Mixed glial cultures were used to demonstrate oligodendrocyte’s extreme vulnerability to the toxic effects of ethanol compared with other cell types. The pro-death effects of ethanol are mediated by inhibition of PI3K and Erk MAPK[24]. Ethanol impaired signaling through PI3K/Akt is due to reductions in insulin/IGF receptor binding[25] associated with altered membrane lipid composition[25] and fluidity[26], as well as decreased phosphorylation and activation of downstream pathways[27-31].
Despite evidence that ethanol exposures can be sufficient to cause FASD, variability in the nature and severity of the phenotype suggests that co-factors may contribute to its pathogenesis. Of note is that a very high percentage of heavy drinkers (up to 80%) also abuse tobacco products, mainly by smoking cigarettes[32]. Review of an outpatient substance abuse treatment center database for pregnant women revealed that from 2010 to 2013, 74% of the pregnant alcohol users (N = 57) smoked cigarettes compared with 42% of controls (N = 31) (P = 0.0053)[33]. Despite strong evidence linking cigarette smoking during pregnancy to impaired fetal growth and development, and neurocognitive function[34], the mechanisms are not well understood. Given the frequency of overlapping exposures and their known independent adverse effects on development, further research is needed to determine the degree to which alcohol and tobacco exposures produce differential or additive adverse effects on white matter in the immature brain.
Although the vast majority of research designed to investigate alcohol-tobacco interactive effects has been focused on carcinogenesis[35-38], particularly in relation to the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and its metabolites[36], it is known that low, sub-mutagenic exposures to nitrosamines such as streptozotocin, N-nitrosodimethylamine (NDEA), and NNK cause insulin resistance with impaired signaling through PI3K-Akt, increased DNA damage, lipid peroxidation, mitochondrial dysfunction, and ER stress[39-42]. The present study tests the hypothesis that sub-mutagenic exposures to NNK are sufficient to cause white matter molecular pathology similar to the effects of ethanol, and possibly exacerbate the adverse effects of ethanol with respect to impairments in oligodendrocyte and neuroglial gene expression.
Methods
Long Evans rat pups were divided into 4 groups and administered 50 μl i.p. injections of: saline vehicle as control; pharmaceutical grade ethanol (2 g/kg in saline); NNK (2 mg/kg in saline); and ethanol + NNK. Ethanol treatments (binge) were administered on postnatal days (P) 2, 4, 6, and 8[43-45], and NNK was administered on P3, P5, P7, and P9. These models simulated 3rd trimester-equivalent human pregnancy exposures to alcohol and tobacco toxins. The rats were sacrificed at 6 weeks of age to examine long-term effects on temporal lobe insulin/IGF-1 signaling through Akt growth and metabolic pathways during late adolescence. All experiments were performed in accordance with protocols approved by Institutional Animal Care and Use Committee at the Lifespan-Rhode Island Hospital, and they conformed to guidelines established by the National Institutes of Health.
Targeted quantitative RT-PCR arrays were used to measure expression of genes linked to oligodendrocyte and astrocyte function (“S Table 1”). The objective was to evaluate ethanol and NNK effects on genes relevant to CNS myelin and white matter development and integrity. PCR primer pairs were designed with MacVector 12 (Cary, NC) and Primer 3 (http://primer3.sourceforge.net/) software (“S Table 2”). Targeted arrays were constructed by spotting and drying primer pairs (100 nmol/5 μl) into individual wells of a Light cycler 480 Multi-well Plate 96 (Roche, Indianapolis, IN). The arrays were sealed and stored at -80°C. On the day of use, the array plates were thawed on ice and 20 μl of reaction cocktail containing cDNA from 5 μg RNA template and Sybr green master mix were added to each well. PCR reactions were performed in a Roche Lightcycler 480 System. Gene expression was analyzed using the ΔΔCt method with results normalized to housekeeping control genes (hypoxanthine-guanine phosphoribosyltransferase, HPRT; and Ribonuclear protein, RNP).
Each experimental group included 8 - 10 rats. Inter-group comparisons were made using two-way analysis of variance (ANOVA) with Tukey or linear trend post hoc tests (GraphPad Prism 6, San Diego, CA). F-ratios and P-values are tabulated. Significant post-test differences and trends (0.05 < P < 0.10) are shown in the graphs. Heat maps were constructed using Version 3.1 of R software. Exploratory analysis identified missing values verified the quality of observed data. To scale the data, row means were subtracted from each cell. The resulting values were further divided by the standard deviation in order to obtain a z-score of each individual cell. Row values are scaled to have a mean = 0 and standard deviation (S.D.) = 1. The resulting values were plotted using a cosmetically modified heatmap library function in R and a 6-color palette. We also applied hierarchical clustering algorithm using Euclidean distance function on the overall table to display a dendrogram of mRNAs.
Results
PCR Targeted Array Studies: PCR arrays were generated to measure expression of 84 genes involved in glial and neuronal growth, maturation, and function. Pilot screening enabled us to eliminate 55 genes due to their low levels of expression or absent inter-group differences. The follow-up studies were focused on 29 genes (Supplementary Tables 1 and 2) in which mRNA levels were measured by quantitative real time PCR using the SA Biosciences protocol (Qiagen, Carlsbad, CA). Gene expression was calculated using the ΔΔ-Ct method with results normalized to HPRT. Data were analyzed by two-way ANOVA with the post hoc Tukey multiple comparisons test. In addition, functionally clustered summary results are depicted in hierarchical heatmaps that were generated in R.
Oligodendroglial Genes: As markers of immature oligodendroglia, we measured nestin, vimentin, PROM1, CNP, PDGFR-α, GC, and GalC, and for mature myelinating oligodendroglia, we measured PLP, MOG, MAG-1, MBP, RTN4, RPAIN, and ST8sia1 expression (Table 1). Significant ethanol effects were observed with respect to PROM1, GC, GalC, MAG-1, RPAIN, and ST8sia1, and trend effects were observed for nestin and PLP. Significant NNK effects were detected for vimentin, PROM1, PDGFR-α, GC, GalC, PLP, MOG, MBP, RTN4, RPAIN, and ST8SIa1, and trend effects occurred for CNP and MAG-1 expression. Significant ethanol x NNK interactive effects occurred for PROM1, GalC, PLP, MBP, and RPAIN, and trend effects were observed for GC.
Graphed results and post hoc repeated measures tests revealed significant intergroup differences with respect to all immature and mature myelin-associated genes except for CNP (Figure 1D). Ethanol’s significant effects were restricted to one immature (GalC) (Figure 1G) and two mature i.e. PLP (Figure 2A) and MAG-1 (Figure 2C) genes that were all increased relative to control. However, there were also statistical trend-wise increases in MOG (Figure 2B) and reductions in MBP (Figure 2D) relative to control.
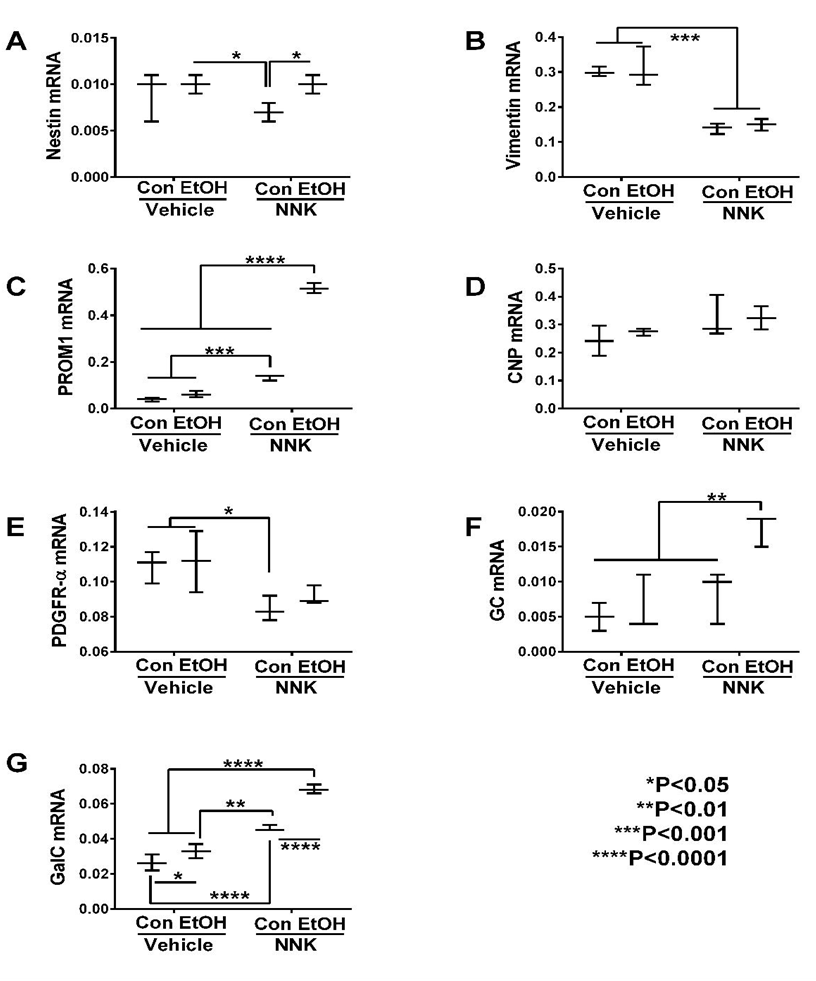
Figure 1: Ethanol and NNK effects on temporal lobe expression of immature oligodendroglial genes. mRNA levels of (A) Nestin, (B) Vimentin, (C) PROM1, (D) CNP, (E) PDGFR-α (F) GC, and (G) GalC, were measured using targeted PCR arrays. Gene expression was calculated using the ΔΔCt method with results normalized to HPRT1. Inter-group comparisons were made by two-way ANOVA (Table 1) with the post-hoc Tukey multiple comparison test.
NNK and particularly ethanol + NNK had broader effects on the expression of both immature and mature oligodendroglial genes, and in several instances, combined exposures produced additive or synergistic responses. NNK significantly increased GalC (Figure 1G), PROM1 (Figure 1C), PLP (Figure 2A), MOG (Figure 2B) RTN4 (Figure 2E), and RPAIN (Figure 2F), and had trend–effect increases on MAG-1 (Figure 2C), but significantly decreased nestin (Figure 1A), vimentin (Figure 1B), PDGFR-α (Figure 1E) and MBP (Figure 2D) relative to control, ethanol, or ethanol + NNK. Ethanol + NNK significantly increased GC (Figure 1F), GalC (Figure 1G), PROM1 (Figure 1C), PLP (Figure 2A), MOG (Figure 2B), RTN4 (Figure 2E), RPAIN (Figure 2F) ST8Sia-1 (Figure 2G), and MAG-1 (Figure 2C), and decreased vimentin (Figure 1B) relative to control. Additive or synergistic stimulatory effects of the dual treatments were observed with respect to GC (Figure 1F), GalC (Figure 1G), PROM1 (Figure 1C), RPAIN (Figure 2F) and ST8SIa-1 (Figure 2G).
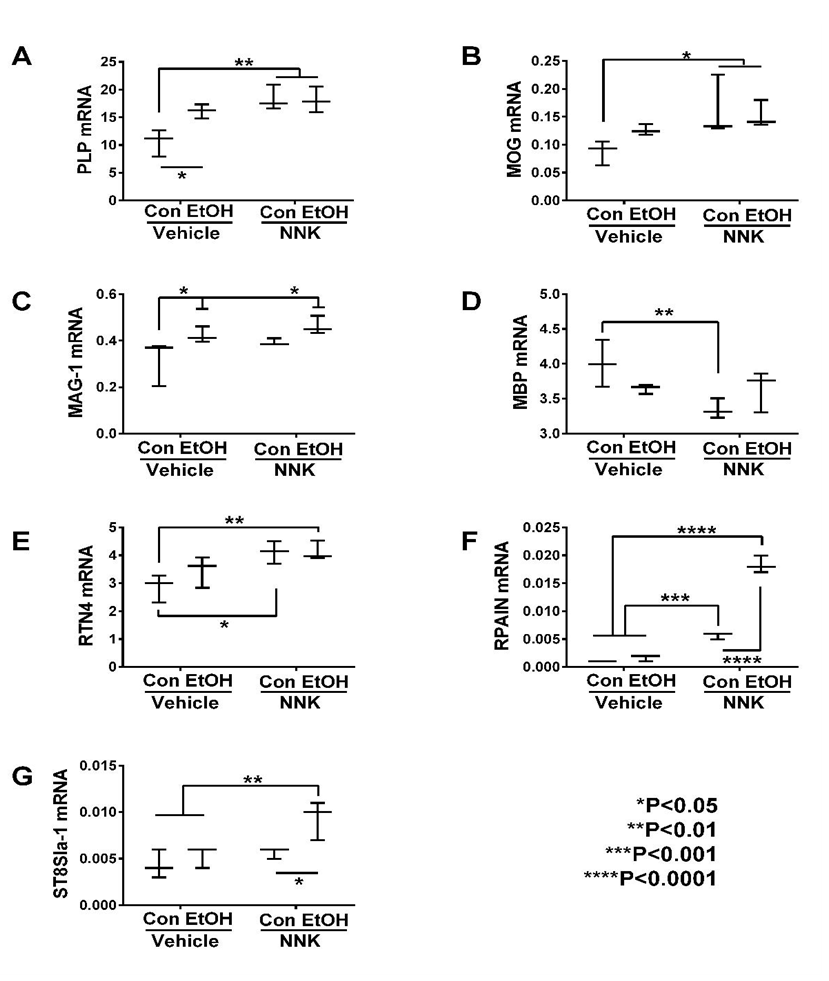
Figure 2: Ethanol and NNK effects on temporal lobe expression of mature oligodendroglial genes. mRNA levels of (A) PLP, (B) MOG, (C) MAG-1, (D) MBP, (E) RTN4, (F) RPAIN, and (G) ST8SIa-1 were measured using targeted PCR arrays. Gene expression was calculated using the ΔΔCt method with results normalized to HPRT1. Inter-group comparisons were made by two-way ANOVA (Table 1) with the post-hoc. Tukey multiple comparisons test.
Oligodendroglial Heatmaps: Heatmaps corresponding to immature and mature oligodendroglial gene expression are depicted in Figure 3. Among the immature oligodendroglial genes, ethanol increased expression of Nestin, CNP, GC, and GalC but decreased PDGR-α. The dual ethanol + NNK treatments produced similar (Nestin, CNP, PDGFR-α), more exaggerated (GC, GAL-C) responses occurred following ethanol + NNK treatments. NNK decreased expression of Nestin and Vimentin, increased PROM1 and CNP, and had no detectable effect on PDGFR-α, GC, or GalC (Figure 3). In essence, ethanol and NNK differentially altered expression of immature oligodendroglial genes, whereas the dual exposures mimicked effects of either or were synergistic (PROM1, GC, GalC).
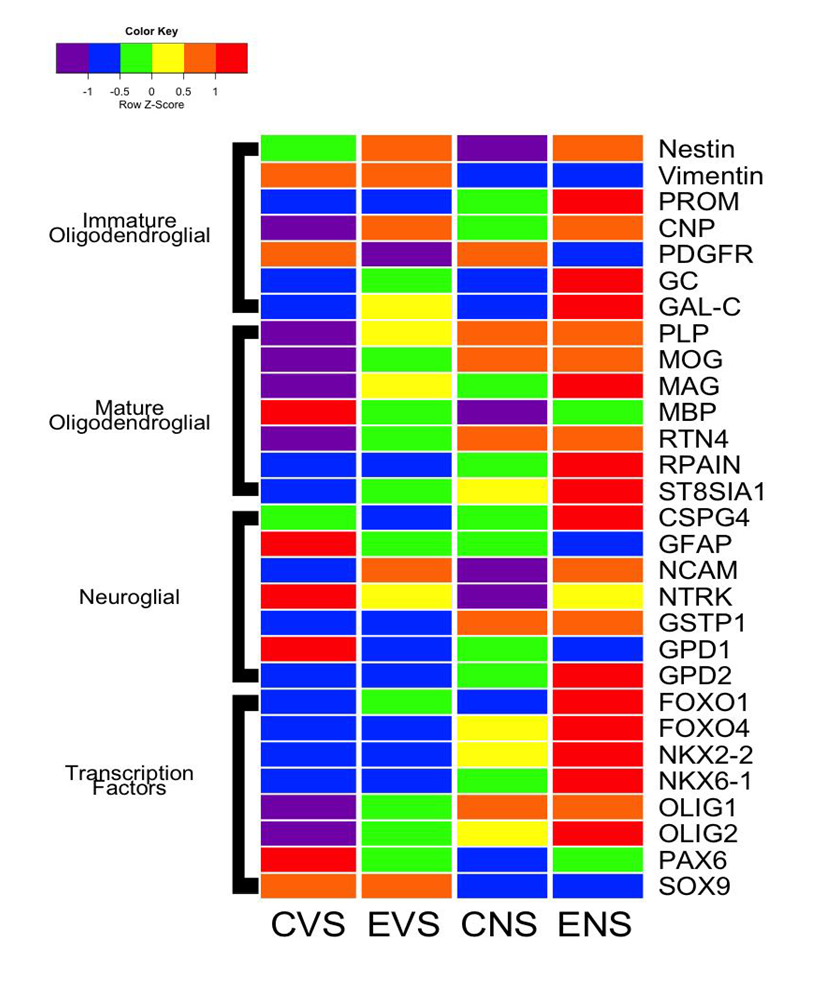
Figure 3: Heatmap with grouping according to gene function. The heatmap was generated using Version 3.1 of R software. Results shown with the 6-color palette correspond to z-scores, which were scaled to have a mean of 0 and S.D. of 1. Results were sorted to juxtapose data corresponding to immature oligodendroglial genes, mature oligodendroglial genes, neuroglial genes, and transcription factors. CVS = control diet with vehicle treatment and saline binge treatment; EVE = ethanol diet with vehicle treatment and ethanol binge; CNS = control diet with NNK treatment and saline binge treatment; ENE = ethanol diet with NNK treatment and ethanol binge.
With regard to mature oligodendroglial genes, ethanol- and NNK-mediated patterns of altered gene expression were more consistent. MBP expression was highest in control brains, and prominently reduced by all other treatments. Otherwise, the expression levels of most genes increased with ethanol, NNK, and ethanol + NNK treatments. For PLP, MOG, RTN4, RPAIN, and ST-8SIA1, gene expression increased from ethanol to NNK. Except for MBP, mature oligodendroglial gene expression was highest in the ethanol + NNK group (MAG, RPAIN, ST8SIA1) or similarly high in the NNK and ethanol + NNK groups (PLP, MOG, RTN4).
Neuroglial Genes: The neural-glial mRNA transcripts measured included: CSPG4, GFAP, NCAM, NTRK2, GSTP1, GPD1 and GPD2. Two-way ANOVA tests demonstrated significant effects of ethanol on GFAP, NCAM, and GPD2, and trend effects on NTRK2 (Table 2). NNK had significant effects on GFAP, NTRK2, GSTP1, and GPD2, and trend effects on CSPG4. Ethanol x NNK interactive effects were significant for NTRK2 and GPD2, and had a trend effect on GFAP. Post hoc tests revealed that ethanol, NNK, and ethanol + NNK significantly reduced temporal lobe expression of GFAP (Figure 4B) and NTRK2 (Figure 4D) relative to control, while NNK and ethanol + NNK increased GSTP1 relative to control and ethanol groups (Figure 4E), ethanol + NNK increased CSPG4 relative to control and ethanol (Figure 4A), and GPD2 relative to the other three groups (Figure 4G). No significant treatment effects occurred with respect to GPD1 (Figure 4F) or NCAM (Figure 4C).
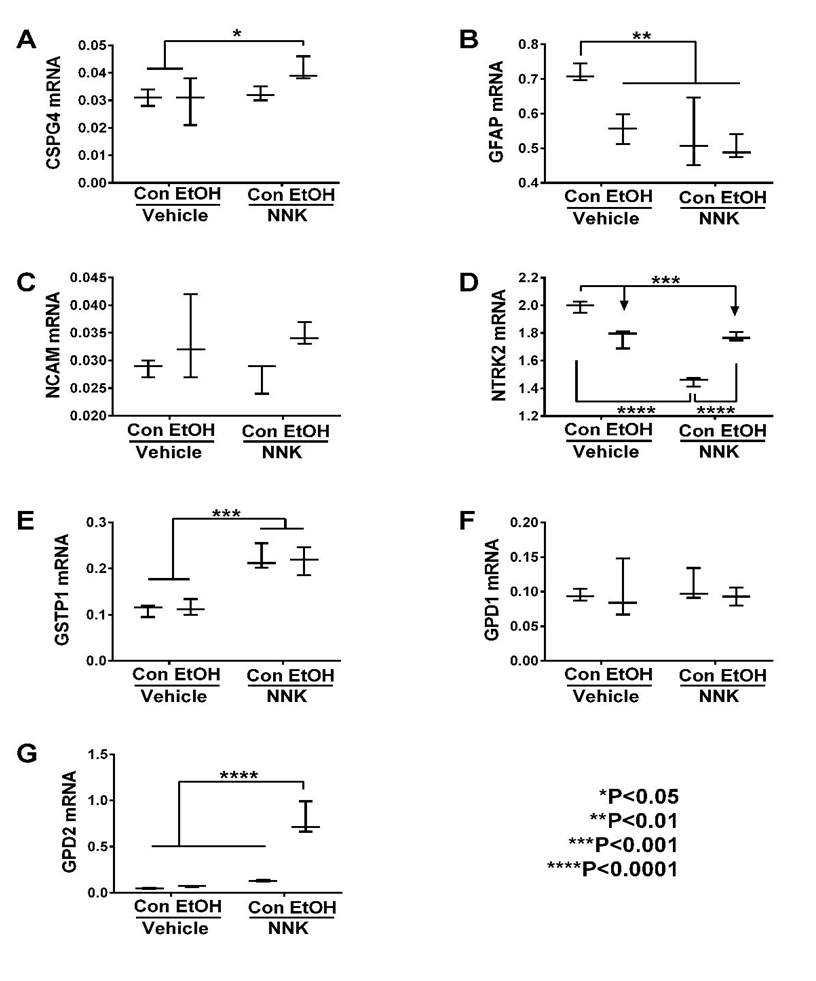
Figure 4: Ethanol and NNK effects on neuronal/glial gene expression in the temporal lobe. (A) CSPG4, (B) GFAP, (C) NCAM, (D) NTKR2, (E) GSTP1, (F) GPD1, and (G) GPD2, mRNA levels were measured using custom targeted PCR arrays. Intergroup comparisons were made by two-way ANOVA (Table 2). Inter-group comparisons were made by two-way ANOVA (Table 2). Tukey post tests assessed differential effects of ethanol and NNK.
Transcription Factors: Glial-associated transcription factor mRNA transcripts measured included: FOXO1, FOXO4, NKX2-2, NKX6-1, Olig1, Olig2, PAX6, and SOX9. Two-way ANOVA tests demonstrated significant ethanol and ethanol x NNK interactive effects on FOXO1, FOXO4, NKX6-1, and PAX6, and NNK effects on all genes except Olig2 (Table 2). Post hoc tests revealed that ethanol significantly increased expression of NKX6-1 (Figure 5D) and decreased PAX6 (Figure 5G) relative to control. NNK significantly increased expression of FOXO4 (Figure 5B), NKX6-1 (Figure 5D) and Olig1 (Figure 5E), and decreased PAX6 (Figure 5G) and SOX9 (Figure 5H) relative to control and ethanol-exposed samples. Ethanol + NNK significantly increased expression FOXO1 (Figure 5A), FOXO4 (Figure 5B), NKX6-1 (Figure 5D), OLIG1 (Figure 5E), and NKX2-2 (Figure 5C), and decreased expression of PAX6 (Figure 5G) and SOX9 (Figure 5H) relative to the control, ethanol, and/or NNK groups. Stepwise increases in gene expression from control or ethanol, to NNK, then ethanol + NNK occurred for FOXO4, NKX6-2, OLIG1, and NKX2-2, and similar trend reductions occurred for PAX6 and SOX9; these responses reflect additive effects of ethanol and NNK in the dual-treatment group.
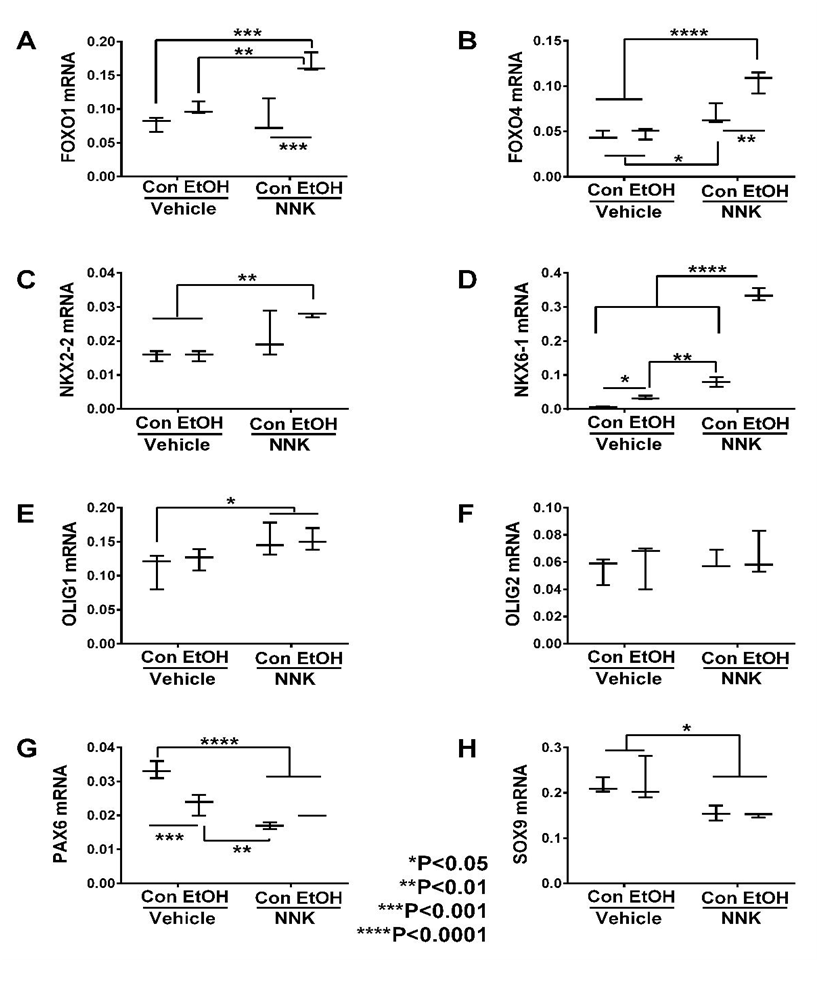
Figure 5: Ethanol and NNK effects on temporal lobe expression of transcription factor genes. The mRNA levels of (A) FOXO1, (B) FOXO4, (C) NKX2-2, (D) NKX6-1, (E) Olig1, (F) Olig2, (G) PAX6, and (H) SOX9 were measured using targeted PCR arrays, and results were calculated using the ΔΔCt method after normalizing to HPRT1. Intergroup comparisons were made by two-way ANOVA (Table 2) with the post-hoc Tukey multiple comparison test.
Neuroglial Heatmap Results: Heatmap analysis demonstrated that control temporal lobes had high mRNA levels of GFAP, NTRK, and GPD1, intermediate levels of CSPG4, and low levels of NCAM, GSTP1, and GPD2 (Figure 3). Ethanol inhibited expression of CSPG4, GFAP, NTRK, and GPD1, maintained low levels of GSTP1 and GPD2, and increased expression of NCAM. NNK’s effects differed entirely from those of ethanol, except that GFAP which was expressed at similar levels. Ethanol + NNK reversed the expression profiles observed in control samples, resulting in lower levels of GFAP, NTRK, and GPD1, and higher levels of CSPG4, NCAM, GSTP1, and GPD2. NNK + ethanol’s effects mimicked responses of ethanol- (NCAM, NTRK and GPD1) or NNK treated (GSTP1) samples, or else they appeared to be synergistic (CSPG4, GFAP, GPD2).
Transcription Factor Heatmap Results: Very low levels of FOXO1, FOXO4, NKX2- 2, NK6-1, Olig1 and Olig2, and high levels of PAX6 and SOX9 in control temporal lobes, and sharply opposite trends in the ethanol + NNK group (Figure 3). Ethanol treatments modestly increased expression of FOXO1, Olig1, and Olig2, reduced expression of PAX6, and caused no change in FOXO4, NKX2-2, NKX6-1, or SOX9 relative to control.
NNK’s effects were intermediate between those of ethanol and ethanol + NNK (FOXO4, NKX2-2, NKX6-1, Olig2), similar to those of ethanol + NNK (Olig1, SOX9), similar to control (FOXO1), or different from the other groups (PAX6).
Discussion
Ethanol and Tobacco Nitrosamine (NNK) Related Brain Disease: This study was designed to examine the independent and interactive effects of ethanol and NNK exposures on neurodegeneration in adult rats. The chronic-binge ethanol exposure model was used to mimic the human clinical scenario that is most prevalent among alcoholics. Chronic low-dose NNK tobacco-specific nitrosamine exposure effects were of interest in relation to alcoholic neurodegeneration because: 1) a very high percentage of heavy drinkers also smoke[32]; 2) the causes of progressive alcoholic brain disease are more complex than alcohol consumption per se; 3) previous studies showed that exposures to sub-mutagenic levels of other nitrosamines cause neurodegeneration[39-41,46]; and 4) we recently showed that early developmental exposures to ethanol and dietary nitrosamines used in preserved and processed foods, i.e. N-nitrosodiethylamine, differentially or additively contributed to cerebellar abnormalities associated with fetal alcohol spectrum disorder[47]. Our working hypothesis herein was that chronic low dose NNK exposures mediate CNS degenerative abnormalities that overlap with and exacerbate adverse effects of alcohol. NNK rather than tobacco smoke effects were studied because smoking could confound the results by causing pulmonary disease[48,49].
Immature oligodendroglial genes: The results show that ethanol had limited effects on the expression of immature oligodendroglial genes and only increased GalC, whereas NNK, with or without ethanol co-exposures significantly modulated expression of 6 of the 7 genes examined. Stimulatory effects occurred for GC, PROM1 and GalC, while inhibitory effects occurred with respect to nestin, Vimentin, and PDGFR-α. Nestin regulates vimentin intermediate filament disassembly during growth and is needed for survival, renewal and proliferation of neural progenitor cells. Inhibition of Nestin corresponds with prior reports of impaired neuronal genesis in chronic ethanol-exposed experimental animals[50,51], illustrating how NNK’s effects can mimic those of ethanol.
PDGFR-β is a cell surface receptor tyrosine kinase expressed in oligoprogenitor cells and when stimulated with PDGF, activates proliferation pathways. Therefore, NNK inhibition would have impaired growth of immature oligodendroglia. GC is a Vitamin D binding nuclear hormone receptor that interacts with RXR and regulates maturation of immature oligodendroglial precursors into myelin-generating oligodendrocytes[52,53]. The significantly elevated levels in ethanol + NNK exposed temporal lobes suggest that the dual treatments may lead to accelerated maturation of oligodendroglia. At the same time, the increased expression of PROM1, particularly in the ethanol + NNK group could have led to suppressed maturation of progenitor or stem cells[54,55], reducing the pool of myelin-generating precursors. GalC (galactosylceramidase/ galactocerebrosidase) hydrolyzes the galactose ester bond of galactosylceramide in its lysosomal catabolism, and either over-expression[56] or deficiency[57] can adversely affect oligodendrocyte maturation and function.
Mature oligodendroglial markers: Gene expression and heatmap analyses of mature oligodendroglial gene expression profiles revealed progressive up-regulation of PLP1, MOG, MAG-1, RTN4, RPAIN, and ST8SIA1 from control, to ethanol, and then NNK exposures, and either further increases with dual exposures, or similar maximum levels of expression as observed in either ethanol or NNK-exposed brains. The generally higher levels of both immature and mature myelin gene expression in ethanol + NNK exposed brains relative to the other groups most likely reflects responses to injury in the early postnatal period. Further studies are needed to understand how these responses impact white matter structure and function in adolescence. MBP was the exception to these trends in that the highest expression levels were in control brains. Ethanol and NNK conspicuously down-regulated MBP relative to control. Since MBP is a major protein constituent of myelin sheaths and has an important role in forming and stabilizing myelin membranes, irrespective of the up-regulation of other myelin genes, MBP’s down regulation would impair the integrity of mature white matter myelin.
Neuroglial genes: The collective responses of neuroglial genes to ethanol and/or NNK exposures were more variably patterned than those observed for oligodendrocyte and transcription factor genes. The reductions in GFAP, NTRK, and GPD1 with ethanol and/or NNK exposures relative to control potentially carry significance with respect to impairments in blood-brain barrier integrity (GFAP), synaptic plasticity (NTRK)[58], and oligodendrocyte responses to stress (GPD1). NNK’s stimulatory, and ethanol + NNK’s further up-regulation of CSPG4 suggests that NNK exposures during development inhibit neurite outgrowth and promote growth cone collapse during axon regeneration[59,60] in adolescent brains, and that these effects are exacerbated by dual exposures to ethanol and NNK. These findings correspond with NNK’s inhibition of NCAM1expression, which mediates neuronal adhesion and neurite outgrowth[58,61]. It is also noteworthy that the ethanol-only effects were opposite those of NNK with respect to these genes, as well as GSTP1 (up-regulated by NNK and not ethanol) which negatively regulates p25-mediated neurodegeneration[62], highlighting the divergent effects of these agent exposures on subsequent CNS molecular pathology. The overall findings with respect to NNK’s effects on CSPG4, NCAM1, and NTRK2 suggest that NNK exposures via tobacco smoke late in development has long-lasting deleterious effects on neuronal plasticity and repair in the adolescent brain.
Transcription factors: The main findings with respect to transcription factors were that the expression levels of FOXO1, FOXO4, NKKX2-2 NKX6-1, Olig1, and Olig2 were increased by ethanol and/or NNK exposures, and for the most part, further sharply increased by ethanol + NNK exposures, reflecting additive or synergistic effects. Opposite effects occurred with respect to PAX6 and SOX9. Since both FOXO1 and FOXO4 target insulin signalling[63-65], and NKX2-2 controls genes that have important roles in axonal guidance[66], the findings suggest that the observed modulations in transcription factor gene expression may represent compensatory efforts to attenuate effects of impaired neuronal growth and plasticity, oxidative stress, and insulin resistance[63,65]. However, the inhibitory effects on PAX6 and SOX9 suggest that ethanol and NNK impair mechanisms of glial progenitor cell maintenance and eventual maturation[67-70], and thereby contribute to white matter pathology in “FASD” that is mediated by developmental exposures to alcohol and/or tobacco smoke (NNK).
Main effects of ethanol and NNK revealed by hierarchical clustered analysis of gene expression. Hierarchical clustering by heatmap analysis revealed broad independent and additive or synergistic effects of ethanol and NNK exposures on temporal lobe gene expression (Figure 6). Two dominant clusters were identified. The upper cluster (a) revealed predominantly low levels of gene expression in control brains and sharply elevated levels in ethanol + NNK exposed brains, whereas the lower dominant cluster (b) had opposite trends. Most of the genes in the lower cluster have important roles in maintaining structural integrity of white matter and synapse integrity in mature brains, whereas most of the genes in the upper cluster participate in various aspects of neuroglial, particularly oligodendrocyte structure and function during development. Concerning the up-regulation of immature neuroglial genes, with the exception of Nestin, NCAM, FOXO1, GC, and GalC, the dominant effects were largely driven by NNK and either maintained or further driven by combined ethanol + NNK exposures. Although the expression of PDGFR-α, SOX9, vimentin, NTRK, MBP, PAX6, GPD1, and GFAP were down-regulated by ethanol and/or NNK, the inhibitor effects were mainly or more prominently caused by NNK, with the exceptions of PDGFR-a and GPD1. Altogether, these analyses highlight the broader effects of late trimester NNK versus ethanol exposures on later development and maturation of white matter in the temporal lobes. In addition, these findings help delineate differential and additive or synergistic effects of developmental ethanol and NNK exposures on white matter structure and function in adolescent brains, and thus begin to explain how these factors contribute to brain pathology and sustained cognitive deficits in FASD.
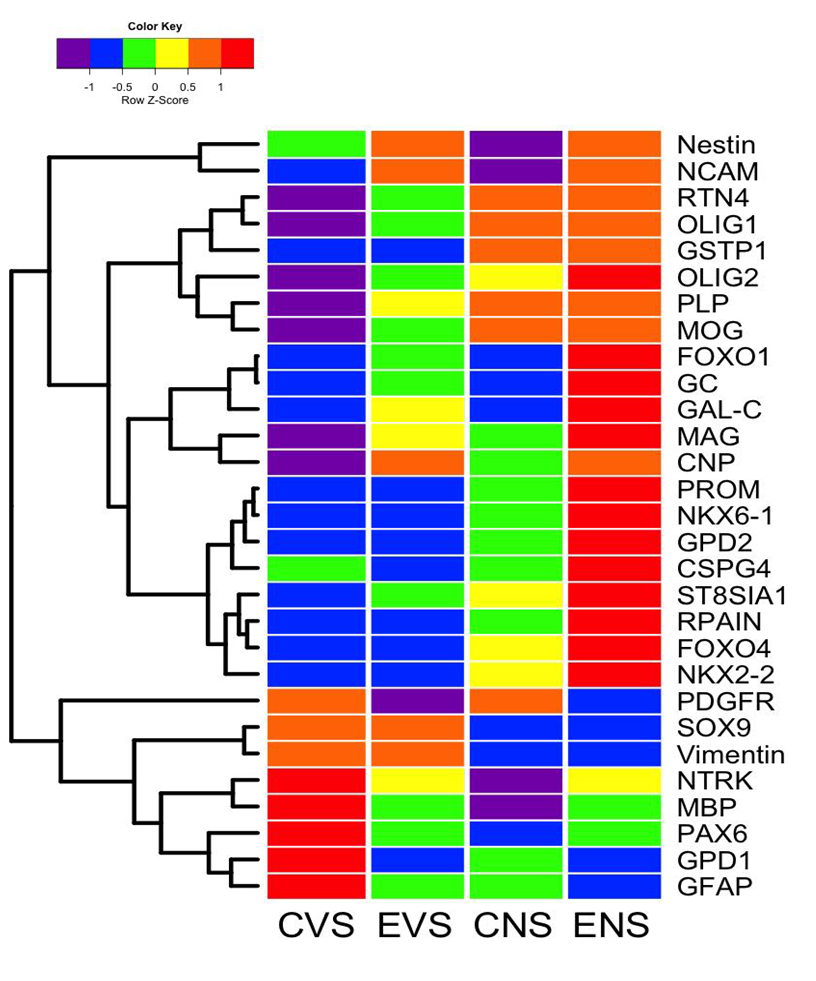
Figure 6: Heatmap illustrating hierarchical clustering of gene expression patterns across experimental groups. The heatmap was generated using Version 3.1 of R software. Results shown with the 6-color palette correspond to z-scores, which were scaled to have a mean of 0 and S.D. of 1. A hierarchical clustering algorithm was applied using the Euclidean distance function on the overall table to display a dendrogram of mRNAs encoding immature oligodendroglial genes, mature oligodendroglial genes, neuroglial genes, and transcription factors (See Figure 3). CVS = control diet with vehicle treatment and saline binge treatment; EVE = ethanol diet with vehicle treatment and ethanol binge; CNS = control diet with NNK treatment and saline binge treatment; ENE = ethanol diet with NNK treatment and ethanol binge.
Supplementary Table 1: PCR Targeted Array Results. Two-way ANOVA Summary of Ethanol and NNK effects on Oligodendroglial Gene Expression.
| Protein | Ethanol Effect | NNK Effect | Ethanol x NNK Effect | |||
|---|---|---|---|---|---|---|
| F- Ratio | P- Ratio | F- Ratio | P- Ratio | F- Ratio | P- Ratio | |
| Immature Oligodendroglia | ||||||
| Nestin | 4.800 | 0.059 | 1.200 | N.S | 1.200 | N.S |
| Vimentin | 0.290 | N.S | 80.18 | < 0.0001 | 0.009 | N.S |
| PROM1 | 576.3 | < 0.0001 | 1065 | < 0.0001 | 457.6 | < 0.0001 |
| CNP | 0.350 | N.S | 4.710 | 0.062 | 0.198 | N.S |
| PDGFR-α | 0.637 | N.S | 12.71 | 0.007 | 0.139 | N.S |
| GC | 8.533 | 0.019 | 16.13 | 0.004 | 4.800 | 0.059 |
| GalC | 55.25 | < 0.0001 | 198.7 | < 0.0001 | 16.12 | 0.004 |
| Mature Oligodendroglia | ||||||
| PLP | 4.670 | 0.063 | 15.63 | 0.004 | 5.512 | 0.047 |
| MOG | 0.614 | N.S | 7.445 | 0.026 | 1.752 | N.S |
| MAG-1 | 7.588 | 0.025 | 3.247 | 0.109 | 0.324 | N.S |
| MBP | 0.066 | N.S | 5.792 | 0.043 | 5.687 | 0.044 |
| RTN4 | 1.345 | N.S | 13.11 | 0.007 | 1.209 | N.S |
| RPAIN | 177.8 | < 0.0001 | 455.1 | < 0.0001 | 144.0 | < 0.0001 |
| ST8sia1 | 7.840 | 0.023 | 10.24 | 0.013 | 2.560 | N.S |
Temporal lobe RNA was analyzed using a targeted PCR array to examine ethanol, NNK and ethanol x NNK interactive effects on Oligodendroglia gene expression. Data were analyzed by Tow-way ANOVA with post-hoc Fisher’s test. Data are graphed in Figure 1 - 3.
Back to Top: ↑
Supplementary Table 2: PCR Targeted Array Results. Two-way ANOVA Summary of Ethanol and NNK effects on Neuroglial and Transcription Factor Gene Expression.
| Protein | Ethanol Effect | NNK Effect | Ethanol x NNK Effect | |||
|---|---|---|---|---|---|---|
| F- Ratio | P- Ratio | F- Ratio | P- Ratio | F- Ratio | P- Ratio | |
| Neuroglial Markers | ||||||
| CSPG4 | 1.642 | N.S | 4.252 | 0.073 | 2.612 | N.S |
| GFAP | 8.237 | 0.021 | 12.18 | 0.008 | 3.518 | 0.098 |
| NCAM | 6.223 | 0.037 | 0.004 | N.S | 0.223 | N.S |
| NTRK2 | 3.366 | 0.104 | 104.5 | < 0.0001 | 111.4 | < 0.0001 |
| GSTP1 | 0.001 | N.S | 63.87 | < 0.0001 | 0.178 | N.S |
| GPD1 | 0.105 | N.S | 0.039 | N.S | 0.421 | N.S |
| GPD2 | 44.38 | 0.0002 | 61.39 | < 0.0001 | 38.59 | 0.0003 |
| Transcription Factors | ||||||
| FOXO1 | 29.79 | 0.0006 | 16.04 | 0.004 | 9.729 | 0.014 |
| FOXO4 | 14.38 | 0.005 | 55.18 | < 0.0001 | 10.83 | 0.011 |
| NKX2-2 | 2.344 | N.S | 18.24 | 0.003 | 2.344 | N.S |
| NKX6-1 | 425.0 | < 0.0001 | 745.4 | < 0.0001 | 277.0 | < 0.0001 |
| Olig1 | 0.432 | N.S | 8.106 | 0.022 | 0.299 | N.S |
| Olig2 | 0.301 | N.S | .590 | N.S | 0.004 | N.S |
| PAX6 | 8.820 | 0.018 | 69.62 | < 0.0001 | 30.42 | 0.0006 |
| SOX9 | 0.021 | N.S | 17.66 | 0.003 | 0.173 | N.S |
Temporal lobe RNA was analyzed using a targeted PCR array to examine ethanol, NNK and ethanol x NNK interactive effects on Oligodendroglial Gene expression. Data were analyzed by Two-way ANOVA with post-hoc Fisher’s test. Data are graphed in Figure 4 - 6.
Back to Top: ↑
References
- 1. Mattson, S.N., Crocker, N., Nguyen, T.T. Fetal alcohol spectrum disorders: neuropsychological and behavioral features. (2011) Neuropsychol rev 21(2): 81-101.
- 2. Riley, E.P., Infante, M.A., Warren, K.R. Fetal alcohol spectrum disorders: an overview. (2011) Neuropsychol rev 21(2): 73-80.
- 3. Bordner, K.A., George, E.D., Carlyle, B.C., et al. Functional genomic and proteomic analysis reveals disruption of myelin-related genes and translation in a mouse model of early life neglect. (2011) Front psychiatry 2:18.
- 4. Nave, K.A. Oligodendrocytes and the "micro brake" of progenitor cell proliferation. (2010) Neuron 65(5): 577-579.
- 5. Goebbels, S., Oltrogge, J.H., Kemper, R., et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. (2010) J Neurosci 30(26): 8953-8964.
- 6. Boulanger, J.J., Messier, C. From precursors to myelinating oligodendrocytes: Contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. (2014) Neuroscience 269: 343-366.
- 7. Campagnoni, A.T., Macklin, W.B. Cellular and molecular aspects of myelin protein gene expression. (1988) Mol neurobiol 2(1): 41-89.
- 8. Gordon, M.N., Kumar, S., Espinosa de los Monteros, A., et al. Developmental regulation of myelin-associated genes in the normal and the myelin deficient mutant rat. (1990) Adv Exp Med Biol 265: 11-22.
- 9. Le Bras, B., Chatzopoulou, E., Heydon, K., et al. Oligodendrocyte development in the embryonic brain: the contribution of the plp lineage. (2005) Int J Dev Biol 49(2-3): 209-220.
- 10. Fancy, S.P., Baranzini, S.E., Zhao, C., et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. (2009) Genes Dev 23(13): 1571-1585.
- 11. Wu, M., Hernandez, M., Shen, S., et al. Differential modulation of the oligodendrocyte transcriptome by sonic hedgehog and bone morphogenetic protein 4 via opposing effects on histone acetylation. (2012) J Neurosci 32(19): 6651-6664.
- 12. Fancy, S.P., Kotter, M.R., Harrington, E.P., et al. Overcoming remyelination failure in multiple sclerosis and other myelin disorders. (2010) Exp neurol 225(1): 18-23.
- 13. He, L., Lu, Q.R. Coordinated control of oligodendrocyte development by extrinsic and intrinsic signaling cues. (2013) Neurosci bull 29(2): 129-143.
- 14. Barres, B.A., Schmid, R., Sendnter, M., et al. Multiple extracellular signals are required for long-term oligodendrocyte survival. (1993) Development 118(1): 283-295.
- 15. Carson, M.J., Behringer, R.R., Brinster, R.L., et al. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. (1993) Neuron 10(4): 729-740.
- 16. Chesik, D., De Keyser, J., Wilczak, N. Insulin-like growth factor system regulates oligodendroglial cell behavior: therapeutic potential in CNS. (2008) J Mol Neurosci 35(1): 81-90.
- 17. Gong, X., Xie, Z., Zuo, H. In vivo insulin deficiency as a potential etiology for demyelinating disease. (2008) Med Hypotheses 71(3): 399-403.
- 18. Ye, P., Kollias, G., D'Ercole, A.J. Insulin-like growth factor-I ameliorates demyelination induced by tumor necrosis factor-alpha in transgenic mice. (2007) J neurosci res 85(4): 712-722.
- 19. D'Ercole, A.J., Ye, P. Expanding the mind: insulin-like growth factor I and brain development. (2008) Endocrinology 149(12): 5958-5962.
- 20. Chiappelli, F., Taylor, A.N., Espinosa de los Monteros, A., et al. Fetal alcohol delays the developmental expression of myelin basic protein and transferrin in rat primary oligodendrocyte cultures. (1991) Int J Dev neurosci 9(1): 67-75.
- 21. Gnaedinger, J.M., Noronha, A.B., Druse, M.J. Myelin gangliosides in developing rats: the influence of maternal ethanol consumption. (1984) J neurochem 42(5): 1281-1285.
- 22. Gnaedinger, J.M., Druse, M.J. Glycoproteins and proteins in an axolemma-enriched fraction and myelin from developing rats: effect of maternal ethanol consumption. (1984) J neurosci Res 12(4): 633-645.
- 23. Kwon, O.S., Schmued, L.C., Slikker, W.Jr. Fumonisin B1 in developing rats alters brain sphinganine levels and myelination. (1997) Neurotoxicology 18(2): 571-579.
- 24. Benjamins, J.A., Nedelkoska, L., Lisak, R.P., et al. Cytokines reduce toxic effects of ethanol on oligodendroglia. (2011) Neurochem Res 36(9): 1677-1686.
- 25. Xu, J., Yeon, J.E., Chang, H., et al. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. (2003) J Biol Chem 278(29): 26929-26937.
- 26. Wing, D.R., Harvey, D.J., Hughes, J., et al. Effects of chronic ethanol administration on the composition of membrane lipids in the mouse. (1982) Biochem Pharmacol 31(21): 3431-3439.
- 27. de la Monte, S., Derdak, Z., Wands, J.R. Alcohol, insulin resistance and the liverbrain axis. (2012) J Gastroenterol Hepatol 27 Suppl 2: 33-41.
- 28. de la Monte, S.M., Wands, J.R. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. (2005)J Alzheimers Dis 7(1): 45-61.
- 29. de la Monte, S.M., Tong, M., Cohen, A.C., et al. Insulin and insulin-like growth factor resistance in alcoholic neurodegeneration. (2008) Alcohol Clin Exp Res 32(9): 1630-1644.
- 30. Ramirez, T., Longato, L., Dostalek, M., et al. Insulin resistance, ceramide accumulation and endoplasmic reticulum stress in experimental chronic alcohol-induced steatohepatitis. (2013) Alcohol Alcohol 48(1): 39-52.
- 31. Ramirez, T., Tong, M., de la Monte, S.M. Chronic-Binge Model of Alcoholic Hepatitis in Long Evans Rats. (2014) Journal of Drug and Alcohol Research 3(2014): 1-10.
- 32. Romberger, D.J., Grant, K. Alcohol consumption and smoking status: the role of smoking cessation. (2004) Biomed pharmacother 58(2): 77-83.
- 33. Tai, M., Piskorski, A., Kao, J.C., et al. Gestational alcohol effects on placental morphology. (2014) Pediatr Dev Pathol 17(2): 146.
- 34. Jacobsen, L.K., Mencl, W.E., Constable, R.T., et al. Impact of smoking abstinence on working memory neurocircuitry in adolescent daily tobacco smokers. (2007) Psychopharmacology 193(4): 557-566.
- 35. de Boer, M.F., Sanderson, R.J., Damhuis, R,A., et al. The effects of alcohol and smoking upon the age, anatomic sites and stage in the development of cancer of the oral cavity and oropharynx in females in the south west Netherlands. (1997) Eur Arch Otorhinolaryngol 254(4): 177-179.
- 36. Duell, E.J. Epidemiology and potential mechanisms of tobacco smoking and heavy alcohol consumption in pancreatic cancer. (2012) Mol carcinog 51(1): 40-52.
- 37. Johnson, N.W., Warnakulasuriy, S., Tavassoli, M. Hereditary and environmental risk factors; clinical and laboratory risk matters for head and neck, especially oral, cancer and precancer. (1996) Eur J cancer Prev 5(1): 5-17.
- 38. Tramacere, I., Negri, E., Bagnardi, V., et al. A meta-analysis of alcohol drinking and oral and pharyngeal cancers. Part 1: overall results and dose-risk relation. (2010) Oral oncol 46(7): 497-503.
- 39. de la Monte, S.M., Tong, M., Lawton, M., et al. Nitrosamine exposure exacerbates high fat diet-mediated type 2 diabetes mellitus, non-alcoholic steatohepatitis, and neurodegeneration with cognitive impairment. (2009) Mol Neurodegener 4: 54.
- 40. Lester-Coll, N., Rivera, E.J., Soscia, S.J., et al. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. (2006) J Alzheimers Dis 9(1): 13-33.
- 41. Tong, M., Neusner, A., Longato, L., et al. Nitrosamine exposure causes insulin resistance diseases: relevance to type 2 diabetes mellitus, non-alcoholic steatohepatitis, and Alzheimer's disease. (2009) J Alzheimers Dis 17(4): 827-844.
- 42. Zabala, V., Tong, M., Yu, R., et al. Potential contributions of the tobacco nicotine-derived nitrosamine ketone (NNK) in the pathogenesis of steatohepatitis in a chronic plus binge rat model of alcoholic liver disease. (2015) Alcohol Alcohol 50(2): 118-131.
- 43. Li, Z., Zharikova, A., Vaughan, C.H., et al. Intermittent high-dose ethanol exposures increase motivation for operant ethanol selfadministration: possible neurochemical mechanism. (2010) Brain Res 1310: 142-153.
- 44. Silvers, J.M., Tokunaga, S., Mittleman, G., et al. Chronic intermittent ethanol exposure during adolescence reduces the effect of ethanol challenge on hippocampal allopregnanolone levels and Morris water maze task performance. (2006) Alcohol 39(3): 151-158.
- 45. de Licona, H.K., Karacay, B., Mahoney, J., et al. A single exposure to alcohol during brain development induces microencephaly and neuronal losses in genetically susceptible mice, but not in wild type mice. (2009) Neurotoxicology 30(3): 459-470.
- 46. Tong, M., Longato, L., de la Monte, S.M. Early limited nitrosamine exposures exacerbate high fat diet-mediated type 2 diabetes and neurodegeneration. (2010) BMC Endocr Disord 10: 4.
- 47. Andreani, T., Tong, M., de la Monte, S.M. Hotdogs and Beer: Dietary Nitrosamine Exposure Exacerbates Neurodevelopmental Effects of Ethanol in Fetal Alcohol Spectrum Disorder. (2014) JDAR 3(2014): 1-9.
- 48. Gan, G., Hu, R., Dai, A., et al. The role of endoplasmic reticulum stress in emphysema results from cigarette smoke exposure. (2011) Cell Physiol Biochem 28(4): 725-732.
- 49. McCaskill, M.L., Kharbanda, K.K., Tuma, D.J., et al. Hybrid malondialdehyde and acetaldehyde protein adducts form in the lungs of mice exposed to alcohol and cigarette smoke. (2011) Alcohol Clin Exp Res 35(6): 1106-1113.
- 50. Crews, F.T., Nixon, K. Mechanisms of neurodegeneration and regeneration in alcoholism. (2009) Alcohol and alcoholism 44(2): 115-127.
- 51. Singh, A.K., Gupta, S., Jiang, Y., et al. In vitro neurogenesis from neural progenitor cells isolated from the hippocampus region of the brain of adult rats exposed to ethanol during early development through their alcohol-drinking mothers. (2009) Alcohol Alcohol 44(2): 185-198.
- 52. Adams, J.S., Chen, H., Chun, R.F., et al. Novel regulators of vitamin D action and metabolism: Lessons learned at the Los Angeles zoo. (2003) J Cell Biochem 88(2): 308-314.
- 53. Christakos, S., Barletta, F., Huening, M., et al. Vitamin D target proteins: function and regulation. (2003) J Cell Biochem 88(2): 238-244.
- 54. Holmberg Olausson, K., Maire, C.L., Haidar, S., et al. Prominin-1 (CD133) defines both stem and non-stem cell populations in CNS development and gliomas. (2014) PLoS One 9(9): e106694.
- 55. Wang, J., O'Bara, M.A., Pol, S.U., et al. CD133/CD140a-based isolation of distinct human multipotent neural progenitor cells and oligodendrocyte progenitor cells. (2013) Stem Cells Dev 22(15): 2121-2131.
- 56. Costantino-Ceccarini, E., Luddi, A., Volterrani, M., et al. Transduction of cultured oligodendrocytes from normal and twitcher mice by a retroviral vector containing human galactocerebrosidase (GALC) cDNA. (1999) Neurochem Res 24(2): 287-293.
- 57. Hawkins-Salsbury, J.A., Shea, L., Jiang, X., et al. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. (2015) J Neurosci 35(16): 6495-6505.
- 58. Skaper, S.D. Neuronal growth-promoting and inhibitory cues in neuroprotection and neuroregeneration. (2012) Methods Mol Biol 846: 13-22.
- 59. Nash, M., Pribiag, H., Fournier, A.E., et al. Central nervous system regeneration inhibitors and their intracellular substrates. (2009) Molecular neurobiology 40(3): 224-235.
- 60. Ghosh, A., David, S. Neurite growth-inhibitory activity in the adult rat cerebral cortical gray matter. (1997) J Neurobiol 32(7): 671-683.
- 61. Skaper, S.D. Neuronal growth-promoting and inhibitory cues in neuroprotection and neuroregeneration. (2005) Ann N Y Acad Sci 1053:376-385.
- 62. Sun, K.H., Chang, K.H., Clawson, S., et al. Glutathione-S-transferase P1 is a critical regulator of Cdk5 kinase activity. (2011) J Neurochem 118(5): 902-914.
- 63. Webb, A.E., Brunet, A. FOXO transcription factors: key regulators of cellular quality control. (2014) Trends Biochem Sci 39(4): 159-169.
- 64. Valverde, A.M., Fabregat, I., Burks, D.J., et al. IRS-2 mediates the antiapoptotic effect of insulin in neonatal hepatocytes. (2004) Hepatology 40(6): 1285-1294.
- 65. Guo, S., Dunn, S.L., White, M.F. The reciprocal stability of FOXO1 and IRS2 creates a regulatory circuit that controls insulin signaling. (2006) Mol Endocrinol 20(12): 3389-3399.
- 66. Habener, J.F., Kemp, D.M., Thomas, M.K. Minireview: transcriptional regulation in pancreatic development. (2005) Endocrinology 146(3): 1025-1034.
- 67. Zhao, X., Wu, J., Zheng, M., et al. Specification and maintenance of oligodendrocyte precursor cells from neural progenitor cells: involvement of microRNA-7a. (2012) Mol Biol Cell 23(15): 2867-2878.
- 68. Nicolay, D.J., Doucette, J.R., Nazarali, A.J. Early stages of oligodendrocyte development in the embryonic murine spinal cord proceed normally in the absence of Hoxa2. (2004) Glia 48(1): 14-26.
- 69. Finzsch, M., Stolt, C.C., Lommes, P., et al. Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGFreceptor alpha expression. (2008) Development 135(4): 637-646.
- 70. Stolt, C.C., Lommes, P., Sock, E., et al. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. (2003) Genes Dev 17(13): 1677-1689.










