
Process and Molecular Modelling Study of Entecavir Drug-Resistant HBV
Affiliation
Department of Chemistry, IPL research center, Lucknow, India
Corresponding Author
KrishnaSarmaPathy, Department of Chemistry, IPL research center, Lucknow, India, E-mail: drkrishnasarmapathy@yahoo.in
Citation
Krishna, S .P., et al. Process and Molecular Modelling Study of Entecavir Drug-Resistant HBV. (2018) J Pharm Pharmaceutics 5(1): 31- 39.
Copy rights
© 2018 Krishna, S.P. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Abstract
Entecavir is an oral antiviral drug used in the treatment of hepatitis B infection. Entecavir is a guanosine nucleoside analogue with selective activity against hepatitis B virus (HBV), which inhibits reverse transcription, Hepatitis B virus (HBV) is highly endemic in South Africa and across sub-Saharan Africa, where around 8 % of people are chronically infected, and rates of HBV-related liver cancer are some of the highest in the world. Globally, viral hepatitis causes approximately 1.3 million deaths every year more than either malaria or tuberculosis with around 240 million people chronically infected with HBV1. The currently available anti-HBV drugs show potent antiviral activity in patients with chronic hepatitis B; however, the resistance and cross-resistance to the drugs is a major obstacle in long-term treatment. Many studies have been conducted to understand the molecular basis of drug resistance, and the mechanistic characterization and molecular modeling of anti-HBV drugs complexed with HBV RT have been reported. Although the three-dimensional X-ray structure of HBV polymerase is not available, its homology model has been reported using the X-ray structure of HIV RT as a template[1-13]. Even though the homology models may not be accurate due to the low sequence homology between the overall HIV and HBV polymerase, the sequence conservation between the RT domains of HIV and HBV polymerase enables molecular modeling of HBV RT[14]. In particular, the residues around the active site that are responsible for recognizing the template-primer or an incoming nucleoside triphosphate are highly conserved. Nucleoside analogue HBV polymerase inhibitors cause chain termination after incorporation into the growing chain in the active site of HBV polymerase and consequently inhibit viral reverse transcriptase. Thus, the HBV homology model structure based on the crystal structure of HIV polymerase serves as a useful guide for understanding the molecular basis of HBV resistance to drugs.
Introduction
The initial patent on entecavir expired in South Africa in 2011 ZA 1991/07894. Current status available on: http://patentsearch.cipc.co.za/, which should have permitted lower-cost generic competitors to enter the market. However, South Africa granted BMS three additional patents on entecavir that only expire between 2022 and 2026. Two of these patents have lapsed meaning BMS has not paid the renewal fees, and they cannot be enforced while one patent covering a lower dosage form of entecavir remains in force. This patent is currently under litigation in India Basheer S. BMS Hepatitis Patent Invalidated: A Viral Effect for India? http://spicyip.com/2013/02/bms-hepatitis-patent-invalidated-viral.html, but because it is in force in South Africa, generic suppliers may be discouraged from bringing their low-dose products to market. A more recent patent on entecavir has not yet been received or processed by the Patents Office, but it could be filed up until the end of 2014 Patent number: WO/2013/177672. Current status available on pa-tentscope.wipo.int. This patent covers the manufacturing process of entecavir, and is an example of patent ever greening where companies file patents on minor changes to an existing drug to maintain patent protection and block competition. The same patent was recently overturned in the United States for failing to meet the criteria of inventive step. However, in South Africa, since no examination of patent applications occurs, if the patent is filed, it is likely to be granted to BMS[15-18]. So long as BMS maintains a monopoly on entecavir in South Africa, the price is likely to remain high, and entecavir will remain out of reach for those who need it. But the crystalline forms of entecavir and its performances are not researched and reported in the above-mentioned patent. The review relates to analogues of 2′-cyclopentyl deoxyguanosine, especially relates to entecavir, its preparation and the pharmaceutical composition and uses thereof. This method was capable to detect Entecavir and its diastereomeric impurities at a level below 0.009 % with respect to test concentration of 500 μg ml-1 for a 20 μL injection volume. The method has shown good, consistent recoveries for diastereomeric impurities (95 – 105 %). The test solution was found to be stable in the diluent for 48 h. The drug was subjected to stress conditions. The mass balance was found close to 99.5 %. Entecavir also helps to prevent the hepatitis B virus from multiplying and infecting new liver cells, is also indicated for the treatment of chronic hepatitis B in adults with HIV/AIDS infection[19-21].
Discussion
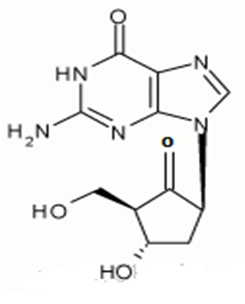
The chemical name for entecavir is 2-amino-I, 9-dihydro-9-[(1S, 3R, 4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl)-6H-purin-6-one, monohydrate. Its molecular formula is C12H15N5O3.H2O, which corresponds to a molecular weight of 295.3 Entecavir, BMS-200475, SQ-34676(1S, 3R, 4S)-9-[4-Hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl] guanine CAS-42217-69-4, 209216-23-9 (monohydrate)
Anti-Hepatitis Virus Drugs, ANTIINFECTIVE THERAPY, Antiviral Drugs -Phase III[22-24]
Patent information
Bristol-Myers Squibb was the original patent holder for Baraclude, the brand name of entecavir in the US and Canada. The drug patent expiration for Baraclude was in 2015. On August 26, 2014, Teva Pharmaceuticals USA gained FDA approval for generic equivalents of Baraclude 0.5 mg and 1 mg tablets; Hetero Labs received such approval on August 21, 2015; and Aurobindo Pharma on August 26, 2015.
Chronic hepatitis B virus infection is one of the most severe liver diseases in morbidity and death rate in the worldwide range. At present, pharmaceuticals for treating chronic hepatitis B (CHB) virus infection are classified to interferon α and nucleoside/nucleotide analogue, i.e. Lamivudine and Adefovir[25,26]. However, these pharmaceuticals cannot meet needs for doctors and patients in treating chronic hepatitis B virus infection because of their respective limitation. Entecavir (ETV) is referred to as 2′-cyclopentyl deoxyguanosine (BMS2000475)[27,28] which belongs to analogues of Guanine nucleotide and is phosphorylated to form an active triple phosphate in vivo. The triple phosphate of entecavir inhibits HBV polymerase by competition with 2′-deoxyguanosine-5′-triphosphate as a nature substrate of HBV polymerase[29-31] , so as to achieve the purpose of effectively treating chronic hepatitis B virus infection and have strong anti-HBV effects [Entecavir, [1S-(1α,3α,4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-hydroxymethyl]-2-methylenecyclopentyl]-6H-purin-6-one, monohydrate, and has the molecular formula of C12H15N5O3.H2O and the molecular weight of 295.3. Its structural formula is as follows:[32,33]
Entecavir was successfully developed by Bristol-Myers Squibb Co. of USA first and the trademark of the product formulation is Baraclude™, including two types of formulations of tablet and oral solution having 0.5 mg and 1 mg of dosage. Chinese publication No. CN1310999 made by COLONNO, Richard, J. et al discloses a low amount of entecavir and uses of the composition containing entecavir in combination with other pharmaceutically active substances for treating hepatitis B[34-44] virus infection, however, the entecavir is non-crystal. In addition, its oral formulations such as tablet and capsule are made by a boiling granulating process. The process is too complicated to control quality of products during humidity heat treatment even though ensuring uniform distribution of the active ingredients.
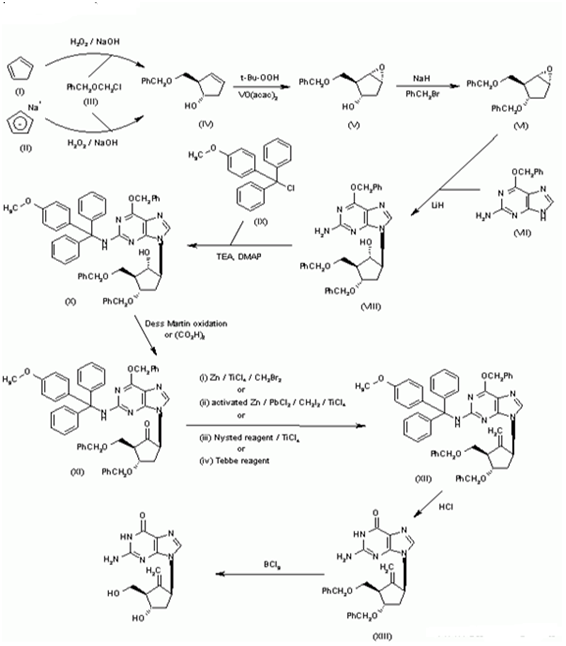
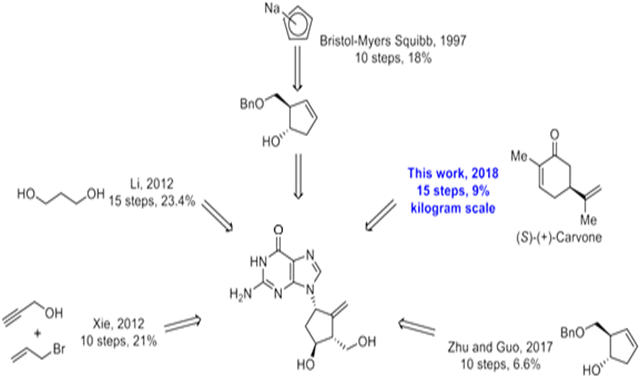
Synthesis of BMS-200475 (EN: 182634) The regioselective reaction of cyclopentadiene (I) and sodium[45] or commercial sodium cyclopentadienide (II)[46,47] with benzyl chloromethyl ether (III) by means of the chiral catalyst (-)-diisopinocampheylborane in THF, followed by hydroxylation with H2O2/NaOH, gives (1S-trans)-2-(benzyloxymethyl)-3-cyclopenten-1-ol (IV), which is regioselectively epoxidized with tert-butyl hydroperoxide and vanadyl acetylacetonate in 2,2,4-trimethylpentane, yielding [1S-(1alpha, 2alpha, 3beta, 5alpha)-2-(benzyloxymethyl)-6-oxabicyclo[3.1.0] hexan-3-ol (V). The protection of (V) with benzyl bromide and NaH affords the corresponding ether (VI), which is condensed with 6-O-benzylguanine (VII) by means of LiH in DMF to give the guanine derivative (VIII). The protection of the amino group of (VIII) with 4-methoxyphenyl(diphenyl) chloromethane (IX), TEA and DMAP in dichloromethane gives intermediate (X), which is oxidized at the free hydroxyl group with methylphosphonic acid, DCC and oxalic acid in DMSO (1) or Dess Martin periodinane in dichloromethane[2,3] yielding the cyclopentanone derivative (XI). The reaction of (XI) with (i) Zn/TiC14/CH2Br2 complex in THF/CH2C12 (1), (ii) activated Zn/PbC12/CH2I2/TiC14 in THF/CH2C12 (2), (iii) Nysted reagent/TiC14 in THF/CH2C12 (2, 3) or (iv) Tebbe reagent in toluene (2) affords the corresponding methylene derivative (XII), which is partially deprotected with 3N HCl in hot THF, providing the dibenzylated compound (XI). Finally, this compound is treated with BC13 in dichloromethane (1-3). (Scheme 18263401a) Description Hydrate, m.p. > 220 C, alpha (22, D) +34? (C 0.3, water) (1); monohydrate, white crystalline solid, m.p. 234-6 C (decomp.) for the bulk sample and m.p. 255 C (decomp.) for an analytical sample recrystallized from water, alpha (D) +33.2? (C 0.3, water) (2); alpha (D) +35.0? (C 0.38, water) (3). Manufacturer Bristol-Myers Squibb Co. (US). Zahler, R., Slusarchyk, W. A. (Bristol-Myers Squibb Co.). Hydroxymethyl (methylenecyclopentyl) purines and pyrimidines. EP 481754, JP 92282373, US 5206244. 2. Bisacchi, G.S., Sundeen, J.E. (Bristol-Myers Squibb Co.). Improved process for preparing the antiviral agent [1S-(1alpha, 3alpha, 4beta)]-2-amino-1, 9-dihydro-9-[4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one.WO 9809964. 3. Bisacchi, G.S., Chao, S.T., Bachard, C. et al. BMS-200475, a novel carboxylic 2’-deoxyguanosine analog with potent and selective anti-hepatitis B virus activity in vitro. Bioorged Chem Lett 1997, 7: 127-32.EP 0481754; JP 1992282373; US 5206244, Zahler, R.; Slusarchyk, W.A. (Bristol-Myers Squibb Co.); Hydroxymethyl (methylenecyclopentyl) purines and pyrimidines. EP 0481754; JP 1992282373; US 5206244, EP 0481754; JP 1992282373; US 5206244, (): WO 9809964.
Preparation method: The reaction steps: a, l is prepared with a short fractionating column (filler may be added) the atmospheric distillation unit, the receiving flask was added anhydrous calcium chloride, and placed in an ice-water bath polymerization (monomer cyclopentadiene rt shall cryopreservation) and tail have to take over the drying tower. Dicyclopentadiene was added to the three-neck flask, the system micro nitrogen, warmed slowly with stirring to 180°C, holding the distillation gas inlet temperature does not exceed 42 °C, to give a final monomer cyclopentadiene (cryopreservation). B
Preparation 2: was added to the kettle in dry xylene, sodium metal to surface oxidation, micro nitrogen, warmed to and stirred at 120 ~ 150°C, the sodium was dissolved, with vigorous stirring, sodium dispersed into sodium sand, stirring was stopped, the system was returned to room temperature, sodium sand cured, removing surface xylene, replacement with a suitable amount of anhydrous THF three times, finally dry THF was added protection, backup. In the micro nitrogen, tetrahydrofuran ice-water bath - Sodium sand cooling 0 ~ 10°C, the prepared cyclopentadiene monomer was slowly added dropwise to a tetrahydrofuran - sodium sand system, control the temperature not exceeding 10°C, After the dropwise addition, the ice-water bath was removed, allowed to warm to rt naturally and stirred for about 3 hours, and sediment sodium is consumed, to give a final solution of sodium cyclopentadiene reddish. C,
Preparation 3: The dimethylphenyl chlorosilane and anhydrous tetrahydrofuran were added to the reaction kettle, micro under N2, and the system was cooled to -70°C or less, dropwise addition of 2, was added dropwise to control the temperature - below 70°C, addition was complete the mixture was stirred at -70°C or less incubated for about 3 hours, TLC the reaction was complete ,, was naturally warmed to 0°C, was slowly added to ice water, stirred, allowed to stand, and the organic phase , washed with saturated sodium bicarbonate solution and extracted with n-hexane with ,, dried over anhydrous sodium sulfate, and concentrated under reduced pressure at 65°C, the final 3 to give a dark yellow oil. d,
preparation of 4: 3 and the reactor was added n-hexane cooled to -10°C, quickly dichloroacetyl chloride dropwise wherein continued stirring, triethylamine and a mixture of n-hexane was slowly added to the system dropwise, the temperature was kept at 5°C or less, addition was complete, the reaction at 0 ~ 4°C for about 4 hours, then warmed to room temperature naturally 8 ~ 10 hours (overnight). Completion of the reaction by TLC. Water was added, stirred for 30 minutes at room temperature, standing layer, extracted with hexane, the organic layers were combined, washed to neutrality with saturated sodium bicarbonate, saturated brine, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 4 dark oil. E, prepared at room temperature for 4 to 5, methanol, water and triethylamine were added to the reaction vessel, stir. Warmed to 75 - 80°C, the reaction 4 ~ 5 hours, the reaction was complete, the system was cooled to below 10°C, potassium carbonate was added, stirred for 30 minutes, was slowly added sodium borohydride (note the paste), slowly raised It was stirred at room temperature for 8 - 10 hours to complete the reaction. At this time, the reaction system PH = 9 - 10. Water was added to the system and quench the reaction, stirred for 0.5 hours, the system was adjusted with concentrated hydrochloric acid to PH = 2 - 3, and extracted with ethyl acetate, the organic phase was separated, washed with saturated brine, and the combined organic phases are dried over anhydrous sodium sulfate, filtered, and concentrated to give a viscous black 5. F,
preparation 6: 5, respectively, and absolute ethanol was added L-amino purified autoclave, stirring chamber for about 1 hour, the crystal precipitation temperature was raised to 50 - 60°C, stirred for 5 - 6 hours, cooled to room temperature stirred for about 3 hours, filtered, the filter cake was washed with small amount of absolute ethanol, the filter cake was dried under reduced pressure at 55°C for 10 hours to give 5 as a light brown powdery solid. (HPLC San 93 %). g, of
Preparation 7: addition of methanol at room temperature and at 6, to ice-salt bath and dry reaction vessel, was added dropwise concentrated sulfuric acid, controlling the temperature below 5°C. After the addition, naturally warmed to room temperature (about 20 - 30°C), reaction was stirred for 10 hours. TLC monitored the reaction. After completion of the reaction, methanol was distilled off under reduced pressure and temperature (process, strict control of the degree of vacuum and temperature, faster), after evaporation to dryness, cooled to room temperature. After addition of ethyl acetate and water, stirred for 5 minutes, and extracted with a separatory funnel, the lower water layer extracted with ethyl acetate, (aqueous layer was put to close the waste water collection tank) the organic layers were combined, washed with saturated aqueous sodium bicarbonate solution to adjust PH to 8 - 9. If the emulsion was filtered with a Buchner funnel, (add a layer of celite on the funnel), and the filtrate fraction with water to a separatory funnel, the upper organic phase was washed twice with saturated brine, dried over anhydrous sodium sulfate, filtered solid sodium sulfate was removed. The organic phase was concentrated under reduced pressure and a temperature of < 50 °C, to give black product was 7. H,
Preparation 8: 7 at room temperature, methanol and water were added to the reaction vessel, stir. Warmed to 75 - 80 °C, 3 to 4 hours of reaction, the reaction was complete, the system was cooled to below 10°C, slowly adding a reducing agent was slowly warmed to room temperature stirred for 6 ~ 7 hours to complete the reaction. Water was added to the system, the reaction was quenched, stirred for 0.5 hours, the system was adjusted with concentrated hydrochloric acid to PH = 5 ~ 6, extracted with ethyl acetate, the organic phase was separated, washed with water, brine, combined organic phases were dried over anhydrous over sodium sulfate, filtered, and concentrated to give a viscous reddish 8.i, 9 are prepared under nitrogen, was added to the reaction vessel and dried in glacial acetic acid and 8, stirred and dissolved, was added boron trifluoride acetic acid, heating up the reaction 5 ~ 15 hours, the solution turned black, the reaction completion, was cooled to room temperature. Methanol was added, the ice bath was added to the reaction flask with 5N potassium hydroxide solution adjusted to PH 7 ~ 9, in this case yellow emulsion was then slowly added dropwise 30% hydrogen peroxide solution, the addition, the ice bath was removed, the under nitrogen, heated up to 70°C incubation for 12 hours. After completion of the reaction, cooled to room temperature, the batch dropwise addition of saturated sodium bisulfite solution. After the addition was stirred for half an hour, then the temperature of the methanol under reduced pressure to recover about 60°C (note the paste, distillation rate is not too fast, methanol and concentrated after a large number of foam generator) the residue was cooled to room temperature, cooled with an ice-salt bath to 0°C, the mixture was adjusted to PH 2 with concentrated hydrochloric acid, as a yellow liquid. Ethyl acetate was added to the reaction flask, stirred for 5 minutes, extracted with ethyl acetate (recyclable apply), if the emulsion serious, can add a little acetone, and the organic layer was dried over anhydrous sodium sulfate, and concentrated to obtain a pale yellow oily liquid 9. J, 9 ketal of
Preparation 10 to give 10 is formed under the action of a ketone. k, prepared at room temperature was added sequentially 3A molecular sieves ll dry dichloromethane and dried to a reaction vessel, under nitrogen, the reaction solution was cooled to about -25°C, was added dropwise(_) - DIPT, dropwise , stirred for 20 minutes at the reaction temperature of about -25°C. Then a solution of Ti (i-oft04, dropwise, stirred at the reaction temperature of about -25°C 20 min; 10 and then added dropwise a mixed solution of dichloromethane, dropwise, the reaction was stirred at temperature of about -25 °C 20 minutes; TBHP solution was then added dropwise, dropwise, stirring was continued at temperature of about -25°C the reaction, the reaction is monitored by TLC developing solvent = petroleum ether: ethyl acetate = 2: 1 (about 4 hours the end of the reaction, this. reaction requires strictly anhydrous, or incomplete reaction) after completion of the reaction, aqueous sodium bisulfate was added dropwise, while the internal temperature does not exceed -10°C, addition was complete, remove the cooling bath was allowed to warm to room temperature Q0-25°C ) The reaction was stirred for 1 hour. Filtration, (add a layer of celite on the funnel), and the filtrate was added water and, after stirring uniformly vibrating, extracted, the aqueous layer extracted twice with dichloromethane top organic phases are combined, washed successively with saturated aqueous sodium bicarbonate and saturated brine washed twice with water, the organic phase is extracted, dried over anhydrous sodium sulfate, filtered to remove solid sodium sulfate. Under reduced pressure to 45 < °C temperature organic phase was concentrated to give 25 g of a pale brown oil 11.
Preparation of 1, 12: successively added at room temperature to the kettle amino-6-benzyloxy guanine, lithium hydroxide monohydrate of DMF and, under nitrogen protection, was heated to 90°C, reaction was stirred for about 16 hours the end of the reaction. TLC monitored the reaction. Developing solvent = petroleum ether: ethyl acetate = 2: 3. After addition of ethyl acetate and saturated brine, stirred for 5 minutes, filtered (diatomaceous add a layer on the funnel) the filtrate was transferred to a separatory funnel and extracted the aqueous layer extracted with ethyl acetate three times below, (the aqueous layer The organic layers were combined waste water collection tank placed close), the upper organic phase was successively washed twice with 50% saturated aqueous citric acid, washed twice with saturated brine, the upper organic phase was dried over anhydrous sodium sulfate, filtered to remove solids sodium. Concentration of the organic temperatures < 65°C, to give brown gum, a crude product 12. Was used directly in the next reaction. M, at room temperature for 13 preparations was added to the kettle in dry dichloromethane, into nitrogen, start stirring. 12 was added, the whole solution was heated to micro (about 30 - 32°C). Under nitrogen, at ice-salt bath was added pyridinium p-toluenesulfonate (PPTS), stirring for 5 minutes the addition was complete, the temperature controlled at 0°C, was added dropwise triethyl orthoformate, addition was complete, the ice bath removed the reaction was stirred for 3 hours with warm water temperature controlled at 25°C. The reaction was monitored by TLC, developing solvent = petroleum ether: ethyl acetate = 2:1. After completion of the reaction, saturated sodium carbonate solution was slowly added, addition was complete the mixture was stirred at 20 - 25°C for half an hour, then added to a separatory funnel and the stationary layers were separated and the lower organic phase was separated; the upper aqueous phase, then with dichloromethane extraction time, the upper aqueous phase into the collection tank. The combined lower organic phases were then washed twice with water; the lower organic phase was dried over anhydrous sodium sulfate, and sodium sulfate to remove solids. The organic phase was concentrated to a temperature ^ 65°C, and then evacuated for 2 hours to give a viscous oil. Under nitrogen above oil was transferred to a 500 ml four-necked flask, stirred and dissolved after acetic anhydride was added, together with acetic acid, an anti- oxidant (BHT) I grains, then dried over anhydrous oxygen-free state, the incubation was heated to 118 - 122°C for about 30 hours. In the course of the reaction incubation, the solution from dark brown to black, TLC the reaction was monitored, after the completion of the reaction, are graded cool. Under nitrogen, when the temperature dropped to 65°C, diluted with water and ethyl acetate were added, the ethyl acetate layer separated, washed with brine, dried over anhydrous sodium sulfate, pouring suspended solids, concentrated, the solid washed with cold ethyl acetate , dried 13 as a white solid. Preparation of [eta], entecavir monohydrate 13, was added 1: 1 THF - methanol, hydrochloric acid was added dropwise 2Ν The reaction was stirred until the starting material was 4. 5h at 60°C, cooled to room temperature, diluted with water and ethyl acetate were added, with vigorous stirring PH was adjusted with iN sodium hydroxide to 7.0, was allowed to stand, a white solid in the organic layer, two phases were separated, the aqueous phase was extracted with ethyl acetate, the combined organic phases were washed with brine, dried over anhydrous sodium sulfate, pouring suspended solids, concentrated solid was washed with cold ethyl acetate, and dried to give entecavir monohydrate as a white solid.
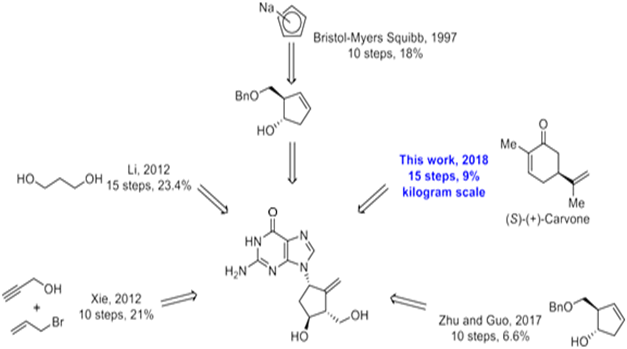
As shown in Scheme 1, compound 3 was prepared as a single diastereomer from 3 kg of 92% ee (S)-(+)-carvone via a two-step transformation including a stereoselective epoxidation and chlorohydrin formation from the newly formed epoxide. Tosylation of the sec-hydroxyl group of compound 3 afforded 4.25 kg of product 4 (60% yield over 3 steps) in 100% ee after recrystallization from MeOH. This ultra-pure intermediate was then reacted with mCPBA to afford epoxide 5, which was converted into diol 6 after treatment with dilute aqueous sulfuric acid. Protection of the diol with dimethoxypropane afforded 3.4 kg of intermediate 7 (67% over 3 steps). This product was treated with sodium methoxide in methanol to initially provide the cis-substituted Favorskii rearrangement product 8a, which upon isomerization gave the thermodynamically more stable cyclopentanecarboxylate 8 under the reaction conditions, though the epimerization wasincomplete even after being stirred for 24 hours (50 g scale) at room temperature. Fortunately, the problem was solved by using methyl t-butyl ether (MTBE)/methanol as the solvent and the reaction was complete in less than 17 hours (50 g scales).
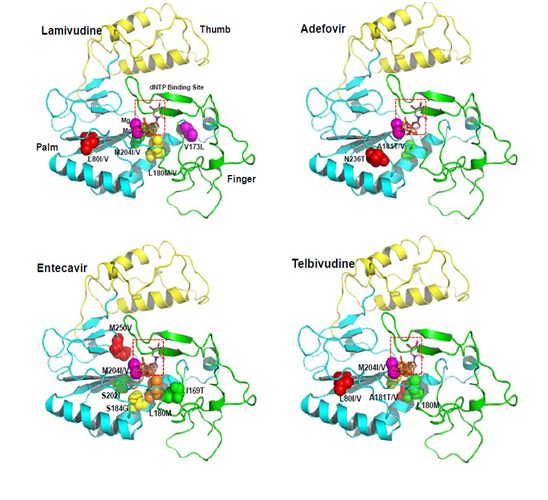
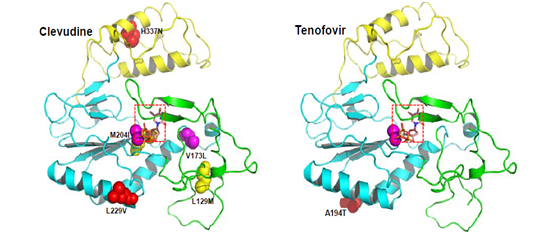
Molecular modeling study of drug-resistant HBV: Molecular dynamics studies on the homology model structure of HBV can provide useful information regarding mutations associated with resistance to inhibitors of HBV polymerase. Daga et al. Built a stereo chemically significant homology model of HBV polymerase and suggested a significant role for conserved Lys 32 residue in HBV RT, which corresponds to Lys 65 in HIV RT, in binding of nucleotides and known HBV RT inhibitors. Their homology model of HBV polymerase had two main differences from previous reports: They aligned the sequence by using the proper match of a conserved Lys residue in HIV-1 RT, which has important salt bridge interactions with the γ-phosphate of the incoming nucleotide. Secondly, they used a different template structure of HIV-1 RT[48-50] (PDB code: 1T05) that was a higher resolution crystal structure compared to the previously.sed template (PDB code: 1RTD). Based on this modeling result, they provided an explanation for the various resistant mutants of HBV polymerase and successfully predicted binding conformations of known HBV inhibitors constructed a three-dimensional homology model of the catalytic core of HBV polymerase based on the crystal structure of HIV-1 RT[16,51]. Molecular modeling studies using the HBV polymerase homology model suggest that steric hindrance between the mutant amino acid side chain and lamivudine or emtricitabine, anti-HBV-drug, could account for the resistance phenotype. Specifically, steric conflict between the Ile or Val at position rt204 in HBV polymerase and the sulfur atom in the oxathiolane ring of lamivudine and emtricitabine is proposed to account for the resistance observed with rtM204I or rtM204V mutation. The effects of the rtL180M mutation, which also occurs near the HBV polymerase active site, appeared to be less direct, potentially involving rearrangement of the deoxynucleoside triphosphate-binding pocket residues. Sharon et al.[5]constructed a homology model structure of HBV polymerase, which is used for minimization, conformational search and induced fit docking followed by binding energy calculation for wild-type and mutant HBV polymerases (rtL180M, rtM204V, rtM204I, rtL180M + rtM204V, rtL180M + rtM204I). Their studies suggest a significant correlation between the fold resistance and the binding affinity of five anti-HBV agents: lamivudine, adefovir, entecavir, telbivudine and clevudine[23,52]. Also, they analyzed different binding modes for synthetic nucleoside analogue drugs as well as natural nucleosides. Although their studies may not fully explain the difference of quantitative binding affinity, they showed detailed resistance mechanisms for anti-HBV drugs against wild-type and mutant HBV. Adefovir is active against wild-type and lamivudine-resistant strains of HBV[7]. In contrast to lamivudine therapy, adefovir is associated with delayed and uncommon selection of drug-resistant viruses[8]. Long-term treatment with adefovir dipivoxil leads to the rtN236T mutation, which displays reduced susceptibility to adefovir but remains sensitive to lamivudine[9]. Yadav V et al[11]. Presented the molecular basis of the mechanism of adefovir-diphosphate against lamivudineresistant mutants and its decrease in susceptibility for rtN236T HBV polymerase mutants. These molecular dynamics studies demonstrated that the rtN236T HBV polymerase mutation does not affect the binding affinity of the natural substrate (dATP), but it decreases the binding affinity of adefovirdiphosphate toward the rtN236T HBV polymerase. The lamivudine-resistant mutations, rtM204V and rtM204I, result in increased van der Waals contacts between adefovir-diphosphate and the mutated residues, which accounts for the better binding affinity of adefovir-diphosphate toward these mutants. The second lamivudine-related mutation, rtL180M, also results in increased van der Waals interactions between adefovir-diphosphate and the final residue of the primer strand, which accounts for the better binding affinity of adefovir-diphosphate in these mutants. Warner et al. determined the prevalence of rtL80V /me mutation in lamivudine-resistant HBV [isolates and characterized the in vitro phenotype of the mutants. Although L80I increases sensitivity to lamivudine and imparts a replication defect, it enhances the in vitro replication of lamivudine-resistant (rtM204I) HBV[9,10]. Molecular modeling revealed that Leu 80 does not interact directly with the enzyme’s substrates. Molecular models of HBV reverse transcriptase showed that, although Leu 80 is located distal to the enzyme’s dNTP binding pocket, substitution of isoleucine for leucine at this site partially restores replication efficiency by sufficiently changing the overall spatial alignment of other residues that are important for catalysis. These results imply that the presence of rtL80I decreases the enzymes, affinity for both dNTPs and lamivudine triphosphate and that the decrease in affinity for lamivudine triphosphate is greater than the decrease in affinity for the natural substrate, dCTP. As mentioned above, using the homology model structure of HBV polymerase, the amino acid changes resulting from mutations that give antiviral resistance can be mapped to functional regions to provide a better understanding of the molecular mechanism of resistance[3-51]. The HBV polymerase consists of four different domains: terminal protein, a space region, a catalytic RT domain and RNase H domain. We constructed and refined the model structure of HBV RT based on the homology to HIV-1 RT according to the reported method[63]. Figure1. The ribbon diagram of homology model structure of HBV RT shows the location of the major mutations that confer resistance to clinically available six drugs. The HBV RT model structure was constructed and refined as previously reported[13]. HBV RT consists of three sub-domains: fingers (amino acid 1 to 49 and 90 to 172, in green), palm (amino acid 50 to 89 and 173 to 267, in cyan), and thumb (amino acid 268 to 351, in yellow). The locations of the mutations are indicated with the sphere model.
Conclusion
Treatment for chronic hepatitis B patients depends on anti-HBV drugs. Even though the currently approved anti-HBV drugs, nucleos(t)ide analogues, show potent and fast antiviral response, several problems remain to be solved. These include the development of resistance and the side effects such as myopathy,[52,60,61] which is induced by mitochondrial damage. Based on advances in the development ofantiviral agents along with newly discovered drug candidates and combination therapy, resistance should not be a great concern in the near future. However, combination therapy for effective control of HBV requires the development of novel drugs that have different mechanisms of action. The mitochondrial damage is mainly due to the high affinity of nucleos(t)ide RT inhibitors for Mitochondrial DNA polymerase gamma. Therefore, all eviation of side effects should be considered in the development of future nucleos(t)ide drugs. In this regard, the fine crystal structure of polymerase gamma was reported recently[62,63]. The elucidation of the polymerase gamma structure establishes a foundation for understanding the molecular basis of the toxicity of anti-retroviral drugs targeting HBV and HIV and the cause of cellular toxicity induced by some antiviral nucleoside analogs. Eventually, these fundamental studies in conjunction with advanced drug development tools will provide valuable information for the development of novel drugs without side effects.New process of Entecavir well defined in patent EP2488522.
References
- 1. Mazzucco, C.E., Hamatake, R.K., Colonno, R.J., et al. Entecavir for treatment of hepatitis B virus displays no in vitro mitochondrial toxicity or DNA polymerase gamma inhibition. (2008) Antimicrob ag chemother 52(2): 598-605.
- 2. Caldecott, K.W. Molecular biology. Ribose--an internal threat to DNA. (2014) Science. 343(6168): 260-261.
- 3. Godon, C., Cordelieres, F.P., Biard, D., et al. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single- strand break repair and radiation susceptibility. (2008) Nucleic Acids Res 36(13): 4454-4464.
- 4. Lin, X., Yuan, Z.H., Wu, L., et al. A single amino acid in the reverse transcriptase domain of hepatitis B virus affects virus replication efficiency. (2001) J Virol 75(23): 11827-11833.
- 5. Sharon, A., Chu, C.K. Understanding the molecular basis of HBV drug resistance by molecular Modeling. (2008) Antiviral Res 80(3): 339-353
- 6. Bartholomeusz, A., Tehan, B.G., Chalmers, D.K. Comparisons of the HBV and HIV polymerase,and antiviral resistance mutations. (2004) Antivir Ther 9(2): 149-160.
- 7. Daga, P.R., Duan, J., Doerksen, R.J. Computational model of hepatitis B virus DNA polymerase: molecular dynamics and docking to understand resistant mutations. (2010) Protein Sci 19(4): 796-807.
- 8. Xiong, X., Flores, C., Yang, H., et al. Mutations in hepatitis B DNA polymerase associated with resistance to lamivudine do not confer resistance to adefovir in vitro. (1998) Hepatology 28(6): 1669-1673.
- 9. Aloman, C., Wands, J.R. Resistance of HBV to adefovir dipivoxil: A case for combination antiviral therapy? (2003) Hepatology 38(6): 1584-1587.
- 10. Dando, T., Plosker, G. Adefovir dipivoxil: A review of its use in chronic hepatitis B. (2003) Drugs 63(20): 2215-2234.
Pubmed||Crossref||Others
- 11. Yadav, V., Chu, C.K. Molecular mechanisms of adefovir sensitivity and resistance in HBVpolymerase mutants: A molecular dynamics study. (2004) Bioorg Med Chem Lett 14(16): 4313-4317.
- 12. Michalak, T.I., Zhang, H., Churchill, N.D., Profound antiviral effect of oral administration of MIV-210 on chronic hepadnaviral infection in awoodchuck model of hepatitis B. (2009) Antimicrob Agents Chemother 53(9): 3803-3814.
- 13. Lee, Y.S., Kennedy, W.D., Yin, Y.W. Structural insight into processive human mitochondrialDNA synthesis and disease related polymerase mutations. (2009) Cell 139(2): 312-324.
- 14. Hsiang, Y.H., Hertzberg, R., Hecht, S., et al. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. (1985) J Biol Chem 260(27): 14873-14878.
- 15. Shim, J.H., Lee, H.C., Kim, K.M., et al. Efficacy of entecavir in treatment-naïve patients with hepatitis B virus-related decompensated cirrhosis. (2010) J Hepatol 52(2): 176-182.
- 16. Danoy, P., Sonoda, E., Lathrop, M., et al. A naturally occurring genetic variant of human XRCC2 (R188H) confers increased resistance to cisplatin-induced DNA damage. (2007) Biochem Biophys Res Commun 352(3): 763-768.
- 17. Nakamura, K., Kogame, T., Oshiumi, H., et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. (2010) PLoS Genet 6(1): 1000828.
- 18. Takata, M., Sasaki, M.S., Sonoda, E., et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. (1998) EMBO J 17(18): 5497-5508.
- 19. Bae, S.H., Yoon, S.K., Jang, J.W., et al. Hepatitis B virus genotype C prevails among chronic carriers of the virus in Korea. (2005) J Korean Med Sci 20(5): 816-820.
- 20. Kim, H., Jee, Y.M., Song, B.C., et al. Molecular epidemiology of hepatitis B virus (HBV) genotypes and serotypes in patients with chronic HBV infection in Korea. (2007) Intervirology 50(1): 52-57.
- 21. Korean Association for the Study of the Liver. KASL clinical practice guidelines: management of chronic hepatitis B. (2012) Clin Mol Hepatol 18:109-162.
Pubmed||Crossref||Others
- 22. Evans, T.J., Yamamoto, K.N., Hirota, K., et al. Mutant cells defective in DNA repair pathways provide a sensitive high Through put assay for genotoxicity. (2010) DNA Repai 9(12): 1292-1298.
- 23. Hochegger, H., Sonoda, E., Takeda, S. Post-replication repair in DT40 cells: translesion polymerases versus recombinases. (2004) Bioessays 26(2): 151-158.
- 24. Broomfield, S., Hryciw, T., Xiao, W. DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. (2001) Mutat Res 486(3): 167-184.
- 25. Chang, T.T., Gish, R.G., de, M. R., et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. (2006) N Engl J Med 354(10): 1001-1010.
- 26. Lai, C.L., Shouval, D., Lok, A.S., et al. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. (2006) N Engl J Med 354(10): 1011-1020.
- 27. Pol, S., Lampertico, P. First-line treatment of chronic hepatitis B with entecavir or tenofovir in ‘real-life’ settings: from clinical trials to clinical practice. (2012) J Viral Hepat 19(6): 377-386.
- 28. Sherman, M., Yurdaydin, C., Sollano, J., et al. Entecavir for treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. (2006) Gastroenterology 130(7): 2039-2049.
- 29. Zoutendijk, R., Reijnders, J.G., Brown, A., et al. Entecavir treatment for chronic hepatitis B: adaptation is not needed for the majority of naive patients with a partial virological response. (2011) Hepatology 54(2): 443-451.
- 30. Manns, M.P., Akarca, U.S, Chang, T.T., et al. Long-term safety and tolerability of entecavir in patients with chronic hepatitis B in the rollover study ETV-901. (2012) Expert Opin Drug Saf 11(3): 361-368.
- 31. Tenney, D.J., Pokornowski, K.A., Rose, R.E., et al. 20 Entecavir maintains a high genetic barrier to HBV resistance through 6 Years in naive patients. (2009) J Hepatol 50(1): 10.
Pubmed||Crossref||Others
- 32. Bristol-Myers Squibb Company. (2005) Entecavir Antivir Dru Adv Co (AVDAC).
Pubmed||Crossref||others
- 33. Study of entecavir in patients with chronic hepatitis B virus infection (2017) NIH.
Pubmed|Crossref|Others
- 34. Tenney, D.J., Rose, R.E., Baldick, C.J., et al. Long-term monitoring shows hepatitis B virus resistance to entecavir ia nucleoside-naive patient is rare through 5 years of therapy. (2009) Hepatology 49(5): 1503-1514.
- 35. Dalmora, S.L., Sangoi, Mda. S., Nogueira, D.R., et al. Validation of a stability-indicating RP-HPLC method for the determination of entecavir in tablet dosage form. (2010) J AOAC Int 93(2): 523-530.
Pubmed||Crossref||Others
- 36. Vijay, A. R., Vinay, K. Ch., Senthil, K. N., et al. (2011) Int J Res Pharm Biomed Sci 2 (3): 1033-40.
Pubmed||Crossref||Others
- 37. Reddy, R., Jamani, S., Suryadevara, V. (2014) Int J Res Ayurveda Pharm 5(4): 531-35.
Pubmed||Crossref||Others
- 38. Asaad, A.M., Al-Ayed, M.S., Aleraky, M., et al. Hepatitis B virus genotyping in chronic hepatitis B patients in southwestern Saudi Arabia. (2015) Braz J infect Dis 19(5): 525-528.
- 39. Martin, P., Lau, D.T., Nguyen, M.H., et al. A Treatment Algorithm for the Management of Chronic Hepatitis B Virus Infection in the United States: 2015 Update. (2015) Clin Gastroenterol and hepatol 13(12): 2071-2087.
- 40. Fung, J., Seto, W.K., Lai, C.L., et al. Extrahepatic effects of nucleoside and nucleotide analogues in chronic hepatitis B treatment. (2014) J Gastroenterol Hepatol 29(3): 428-434.
- 41. World journal of hepatology. (2014) 6(5): 284-92.
Pubmed||Crossref||Others
- 42. Tenney, D.J., Rose, R.E., Baldick, C.J., et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. (2009) Hepatology 49(5): 1503-1514.
- 43. Yang, S.W., Kim, G.H., Chung, J.W., et al. Prediction of risk for hepatocellular carcinoma by response of serum α-fetoprotein to entecavir therapy. (2015) J Gastroenterol Hepatol 30(7): 1175-1182.
- 44. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. (2012) J hepatol 57(1): 167-185.
- 45. HepatitisB. (2015) WHO.
Pubmed||Crossref||Others
- 46. Chae, H.B., Kim, J.H., Kim, J.K., et al. Current status of liver diseases in Korea: hepatitis B. (2009) Korean J Hepatol 15(6):13- 24.
- 47. Cheongju (KR): Korea Centers for Disease Control & Prevention. (2012).
Pubmed||Crossref||Others
- 48. Hou, J.L., Jia, J.D., Wei, L., et al. Efficacy and safety of entecavir treatment in a heterogeneous CHB population from a ‘real world’ clinical practice setting in China. (2013) J Viral Hepat 20(11): 811-820.
- 49. Mazzucco, C.E., Hamatake, R.K., Colonno, R.J., et al. Entecavir for treatment of hepatitis B virus displays no in vitro mitochondrial toxicity or DNA polymerase gamma inhibition. (2008) Antimicrob Agents Chemother 52(2): 598-605.
- 50. Liaw YF, Raptopoulou-Gigi M, Cheinquer H, et al. Efficacy and safety of entecavir versus adefovir in chronic hepatitis B patients with hepatic decompensation: a randomized, open-label study. (2011) Hepatology 54(1): 91-100.
- 51. Matsuzaki, Y., Adachi, N., Koyama, H. Vertebrate cells lacking FEN-1 endonuclease are viable but hypersensitive to methylating agents and H2O2. (2002) Nucleic Acids Res 30(14): 3273-3277.
- 52. Liaw, Y.F., Leung, N., Kao, J.H, et al. Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2008 update. (2008) Hepatol int 2(3): 263-283.
- 53. Lok, A.S., Mc Mahon, B.J. Chronic hepatitis B: update 2009. (2009) Hepatology 50(3): 661-662.
Pubmed||Crossref||Others
- 54. Brambilla, G., Mattioli, F., Robbiano, L., et al. Studies on genotoxicity and carcinogenicity of antibacterial, antiviral, antimalarial and antifungal drugs. (2012) Mutagenesis 27(4): 387-413.
- 55. Friedrich, A., Olejniczak, K. Evaluation of carcinogenicity studies of medicinal products for human use authorised via the European centralised procedure (1995–2009). (2011) Regul toxicolo and pharmacol 60(2): 225-248.
- 56. Bristol-Myers Squibb Co. Baraclude (entecavir). (2010) US prescribing informat.
Pubmed||Crossref||Others
- 57. Brown, J.A., Pack, L.R., Fowler, J.D., et al. Presteady state kinetic investigation of the incorporation of anti-hepatitis B nucleotide analogues catalyzed by noncanonical human DNA polymerases. (2012) Chem Res Toxicol 25(1): 225-233.
- 58. Yamamoto, K.N., Hirota, K., Kono, K., et al. Characterization of environmental chemicals with potential for DNA damage using isogenic DNA repair-deficient chicken DT40 cell lines. (2011) Env mol mutag 52(7): 547-561.
- 59. Adachi, N., So, S., Koyama, H. Loss of Nonhomologous End Joining Confers Camptothecin Resistance in DT40 Cells. (2004) J Biol Chem 279(36): 37343-37348.
- 60. Celeste, A., Fernandez-Capetillo, O., Kruhlak, M.J., et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. (2003) Nat cell boil 5(7): 675-679.
- 61. Innaimo, S.F., Seifer, M., Bisacchi, G.S., et al. Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. (1997) Antimicrob ag chemother 41(7): 1444-1448.
- 62. Caldecott, K.W. DNA single-strand break repair. (2014) Exp cell Res 329(1): 2-8.
Pubmed||Crossref||Others
- 63. Falkenberg, M., Larsson, N.G. Structure casts light on mtDNA replication. (2009) 139(2): 231-233.










