
Structure Based Discovery of inhibitors for Multidrug Efflux Pump- AcrB
Rahul Ramtekkar1, Waheeta Hopper2, M. Michael Gromiha3, Kazuhiko Fukui4D. Velmurugan1,5*
Affiliation
- 1Center of Advanced Study in Crystallography and Biophysics, University of Madras, Maraimalai (Guindy) Campus, Chennai, India
- 2Department of Bioinformatics, School of Bioengineering, Faculty of Engineering and Technology, SRM University, Kattankulathur
- 3Department of Biotechnology, Indian Institute of Technology Madras, Chennai, India
- 4Computational Biology Research Center, National Institute of Advanced Industrial Science and Technology, Tokyo, Japan
- 5Bioinformatics Infrastructure Facility, University of Madras, Maraimalai (Guindy) Campus, Chennai, India
Corresponding Author
Velmurugan, D. Center of Advanced Study in Crystallography and Biophysics, University of Madras, Maraimalai (Guindy) Campus, Chennai – 600 025, India; E-mail: shirai2011@gmail.com
Citation
Velmurugan, D., et al. Structure Based Discovery of inhibitors for Multidrug Efflux Pump-AcrB (2015) Bioinfo Proteom Img Anal 1(2): 27 -35.
Copy rights
© 2015 Velmurugan, D. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Abstract
In Escherischia coli, AcrB is an inner membrane transporter that cooperates with a membrane fusionprotein, AcrA and an outer membrane channel TolC, to export a wide variety of drugs directly out of the cell, bypassing the periplasm. By overproducing such membrane transport proteins, E. coli and other human pathogenic bacteria are able to achieve multidrug resistance. In this paper, we have reported the virtual screening of five in silico small molecule libraries against the substrate recognition site situated in the porter domain of binding protomer of AcrB using tools of Schrödinger suite, which resulted in six potential hits. All these compounds favored hydrogen bonding interactions with mainly polar and aromatic residues such as Ser134, Phe178 and Ile671. From the observed results these ligands are suggested to be potent candidates for inhibiting AcrB. Docking simulations were also carried out with several known substrates and inhibitors in order to study their interactions with the binding site.
Introduction
Modern medicine was transformed with the advent of antibiotics and led to the complacent view that many potentially lethal infections have been taken care for good. At the same time widespread usage of these antibiotics created selective pressure on various bacterial strains for antibiotic resistance[1]. As a result, the potency of existing antibiotics is depleting due to emerging antibiotic resistance mechanisms[2-4]. Antibiotic resistance occurs by three broad mechanisms, namely, antibiotic receptor alteration, antibiotic modification and antibiotic efflux[5-9]. Although the first two mechanisms have been addressed quite successfully in the past by designing better antibiotics and inhibitors of antibiotic modifying enzymes[10], resistance caused by drug efflux in which membrane transport proteins, which are capable of pumping a wide variety of chemically unrelated compounds including antibiotics and chemotherapeutics is an emerging problem for global public health[11-13]. Over-expression of multidrug efflux pumps has been increasingly found to be associated with clinically relevant drug resistance. Active antibiotic efflux was first hypothesized in 1978 as a causative mechanism of resistance to tetracycline’s in Escherichia coli[14-16]. It has since been found to be a widespread mechanism in both gram positive and gram negative organisms to expel antibiotics. Efflux pumps also play a major role in extrusion of poorly diffusible endogenous molecules[17,18] and potentially harmful exogenous diffusible substances[19,20].
Resistance nodulation cell division (RND) transporters such as AcrB and its homologues are major multidrug efflux transporters in gram negative bacteria such as E. coli and Pseudomonas aeruginosa[4]. AcrB cooperates with an outer membrane factor (OMF), TolC and a membrane fusion protein (MFP), AcrA to form a tripartite complex, AcrAB- TolC (Figure 1) which exports a wide variety of antibiotics, antiseptics, anti-cancer chemotherapeutics and toxic compounds including anionic, cationic, zwitter-ionic and neutral compounds directly out of the cell through a mechanism driven by proton motive force PMF)[21,22]. AcrB structure comprises an asymmetric trimer of 1049 residue protomers which exhibits sequence homology and similar structural architecture between its N-terminal and C-terminal halves, indicating an early gene duplication event[23]. Each protomer consists of a transmembrane domain containing 12 trans membrane helices (TM1-TM12), periplasmic or porter domain containing 4 sub domains(PN1, PN2, PC1 and PC2) – each of which consisting of two β-α-β sandwiches and a TolC docking domain containing 2 sub domains, namely, DN and DC[24-27]. AcrB transports drugs by a three-step functionally rotating mechanism in which the periplasmic domain of each protomer assumes unique conformations called access, binding and extrusion in the process of effluxing the substrates[27]. The conformational cycling and functional rotation of the AcrB is reminiscent of the rotational catalysis in F1F0-ATPase[28].
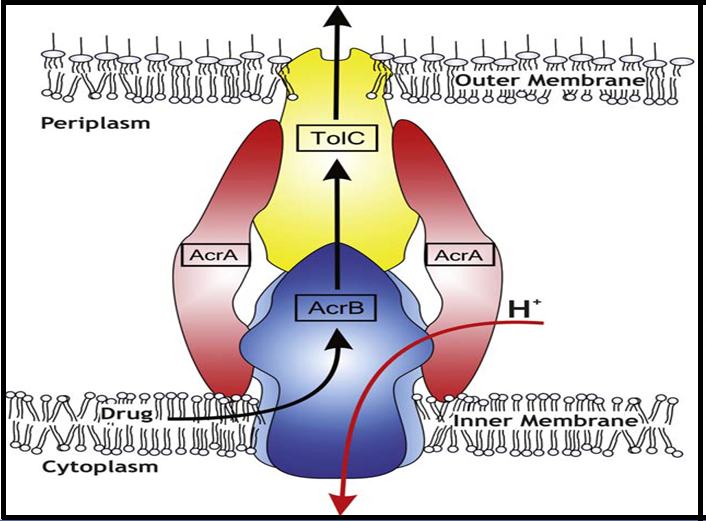
Figure 1: AcrAB-TolC:Schematic drawing of the AcrAB-TolC tripartite efflux pump[47].
In recent years, Molecular Dynamics simulation is also used to study the mechanism of the AcrB and that of the whole tripartite pump. As the structure of AcrB is massive, the AcrB trimer with membrane and solvent can go up to 500,000 atoms, the time scale reachable by conventional all-atom molecular dynamics simulation is far below the time scale relevant to AcrB functional cycle. There are workarounds for this problem such as structure-based coarse-grained model for simulations, targeted molecular dynamics, etc. Yao[29] used structure-based coarsegrained model of AcrB to study the functional rotating mechanism and found indications of substrate extrusion followed by conformational change and stabilization. Substrate bound asymmetric structure was stabilized via allosteric coupling. Schulz[30] and Feng[31] used Targeted Molecular simulations to mimic functional rotation of AcrB containing substrates to understand how the substrate behaves and also the way the binding pocket behaves. They found the substrates to be pushed forward towards extrusion and the conformational changes towards closing of the binding pockets.
The efflux mechanisms responsible for the decreased effectiveness of common antibiotics also accounts for resistance to new and recently described antimicrobial agents[32]. Piddock[33] described patients with salmonellosis who did not respond to ciprofloxacin therapy which was just introduced into clinical use and was considered as immune to efflux resistance as it was synthetic and therefore completely novel to the bacteria. This strongly warrants the developments of compounds that are able to circumvent or block efflux pumps in order to restore antibacterial potency of older as well as newer antibiotics[34,35]. There are a few inhibitors to AcrB such as Phenyl-Arginine-beta- napthylamide (PAβN) and derivatives[36], and 1-(1-naphthylmethyl)- piperazine (NMP)[37], but none of the inhibitors is in advanced stages of drug development. The substrate recognition site of AcrB is in the porter domain of the binding protomer. Two co-crystal complexes of AcrB with its substrates minocycline and doxorubicin in the substrate recognition site have been determined[25] which can be used to perform molecular docking experiments with the aim of finding new ligands to the binding site. In this approach, ligands are computationally docked into the three dimensional structure of the target site. Scoring is performed based on the complementarily and interactions of the ligand with the binding site. This allows ranking of libraries of compounds screened according to their calculated interaction scores and energy. Nikaido[25] have used computational docking simulations to classify the various substrates into two classes namely cave binders and groove binders based on the location of the compounds in the large binding site. Such methods have been successful in efflux pumps of other microorganisms such as S. aureus and P. aeruginosa[39,40]. Docking simulations are performed with several substrates of AcrB to study their interactions with the substrate recognition site. Few in silico libraries were computationally screened for activity against AcrB. Among all the available crystal structures of AcrB, 2J8S[27] was selected due to its high resolution (2.54 Å).
Methods
Ligand preparation
The structures of 14 known substrates and two inhibitors were drawn in SDF format using the tool Marvin Sketch [Marvin Draw 5.1.5, 2008, Chemaxon Ltd., Budapest]. Aromatic scaffolds of Phe-Arg-β-napthylamide were replaced with aliphatic chains of varying lengths and screened against Pubchem small molecule database[41] for 60% similar compounds which resulted in 63000 compounds. Similarly, the Pubchem database was also screened for compounds similar to NMP which generated 27000 compounds. Three openly available small molecules databases, namely, Asinex database (384000 compounds), TOS Lab database (15000 compounds) and May bridge Hitfinder (24000 compounds) were also used in the study. The ligands were prepared for docking simulations using the LigPrep module of the Schrödinger suite of tools. First, all the hydrogen atoms were added to the ligand molecules as they had implicit hydrogen atoms. The bond orders of these ligands were fixed. The ionization states of the ligands were generated in the pH range of 5.0 to 9.0 using epik[42], as the micro-environment of the binding site can fluctuate over these pH values. Most probable tautomers and all possible stereo isomers were generated to study the activity of individual stereotypes of each ligand. In the final stage of LigPrep, compounds were minimized with OPLS-2001 Force field[43].
Protein Preparation
Three protein structures belonging to PDBID 2J8S (Resolution- 2.54 Å), 2DRD (Resolution- 3.30 Å, Co-crystal complex with minocycline) and 2DR6 (Resolution- 3.10 Å, Co-crystal complex with doxorubicin) were downloaded from Protein Data Bank. The complete asymmetric trimer cannot be processed due its size, therefore only the binding protomer was used for docking as it contains the substrate recognition site. Protein preparation plays a very important role in the in silico docking simulation studies as the correctness of the protein structure is crucial to get the correct interactions with the ligands. The protein structures were prepared by a multi-step process through the ‘Protein Preparation Wizard’ of the Schrödinger suite. Firstly, the bond orders in the proteins were assigned, hydrogen atoms were added and all the crystallographic water molecules were removed. The ligands in 2DRD and 2DR6 were retained for the purpose of grid generation. The structures were then subjected to single-point energy calculation using the protein modeling package, Prime. This calculation was carried out using OPLS-2001 force field incorporating implicit solvation.
Following the above steps of preparation, the protein-ligand complex was subjected to energy minimization using the Schrödinger implementation of OPLS-2005 force field with implicit solvation. The entire complex was minimized and the minimization terminated when the root mean square deviation (RMSD) of the heavy atoms in the minimized structure relative to the X-ray structure exceeded 0.3 Å. This helps to maintain the integrity of the prepared structure relative to the experimental structure, while eliminating bad contacts between atoms. Prepared protein was analyzed using the SiteMap [SiteMap, version 2.3, Schrödinger, LLC, New York, NY, 2009] module of the Schrödinger suite of tools. The sitemap gives an idea about which positions are favorable for a donor, accepter or hydrophobic group to be present in the receptor.
Semi-Flexible Docking studies Docking studies on prepared ligands were carried out in the substrate recognition site in B Chain of 2J8S using the docking program, Glide [Glide, version 5.5, Schrödinger, LLC, New York, NY, 2009]. The shape and properties of the receptor were represented on a grid by several different sets of fields that help progressively in more accurate scoring of the ligand poses. The protein-ligand complexes prepared as described above were employed to build energy grids using the default values of protein atom scaling (1.0) within a cubic box dimensions 36 Å × 36 Å × 36 Å centered the residues of the site. The bounding box dimensions (within which the centroid of a docked pose is confined) were set to 14 Å × 14 Å × 14 Å.
In this docking simulation, semi-flexible docking protocols were used. The ligands being docked were kept flexible, in order to explore an arbitrary number of torsional degrees of freedom in addition to the six spatial degrees of freedom spanned by the translational and rotational parameters. The ligand poses generated were passed through a series of hierarchical filters that evaluated the ligand interactions with the receptor. The process of virtual screening was carried in three phases, using three different protocols i.e. High-Throughput Virtual Screening (HTVS), Standard Precision (SP) and Extra Precision (XP) docking protocols. All the tautomeric and isomeric duplications were removed from the ligands selected using virtual screening.
Induced Fit Docking and QPLD Studies Induced fit docking studies were carried out on the selected ligands from the semi-flexible docking studies, wherein induced fit models have been obtained to fit ligands in non-cognate structures. In other words the protein structure was induced to fit the ligands. The procedure described by Sherman[44] was executed via a python script in the framework of Maestro v9.0.109. For the protein model, initial docking was performed with a grid (defined using the centroid of the residues of the site) with default parameters. Twenty poses were chosen to be saved after initial Glide docking, which was carried out with the van der Waals scaling of 0.4 for both protein and ligand non-polar atoms. After obtaining initial docking poses, Prime side chain and backbone refinement together with the minimization of the docked pose was carried out within a sphere of 5Å from each pose saved. Glide re-docking was carried out in Prime refined structures having Prime energy values within 20 kcal/mol of the lowest energy value. The RMSD values of ligand poses having best induced fit docking (IFD) score were determined.
QM-Polarized Ligand Docking (QPLD) is a combination of Glide and QSite, which uses ab initio methodology to calculate ligand charges within the protein environment and therefore offers substantially enhanced accuracy over pure Molecular Mechanics (MM) docking algorithms. Although MM force fields are capable of modeling partial atomic charges on ligands with reasonable accuracy, they are generally incapable of considering charge polarization induced by the protein environment[45]. All the ligands selected after XP docking were processed through QPLD for better accuracy.
Results and Discussion
The Substrate Recognition Site The conformational cycling and functional rotation mechanism of the AcrB have two key activities which are essential for the AcrAB-TolC system to function: energy transduction and substrate recognition. As AcrB is energized by proton-motive force, transient protonation of titratable groups within the trans membrane domain of the protein (i.e. energy transduction) provides the energy for the conformational change from binding to extrusion protomer. Site-directed mutagenesis studies on AcrB revealed that four residues located in the middle of TM4 and TM10, Asp 407, Asp408, Lys 940 and Arg971 form ion pairs and are essential for the functioning of the protein[25]. Substrate recognition takes place in a large substrate binding site which exists in the porter domain of protomer and branches off a large tunnel that putatively functions for drug transport (Figure 2). The site is rich in aromatic amino acids such as Phe136, Phe18, Phe610, Phe615, Phe617 and Phe628 which can form hydrophobic or aromatic-aromatic interactions with the substrate and drug molecules. Two co-crystal structures (2DRD and 2DR6) show minocycline and doxorubicin bound to one of the three protomers (binding conformation) at the distal end of the binding pocket between the β-sheets of PN2 and PC1. This substrate recognition site was used to perform molecular docking experiments with the aim of finding new ligands which can have better interactions with the site. The large binding pocket has been divided into two comparatively small areas, namely, groove and cave[46]. The structure selected for the purpose was 2J8S because of better resolution of 2.54 Å.
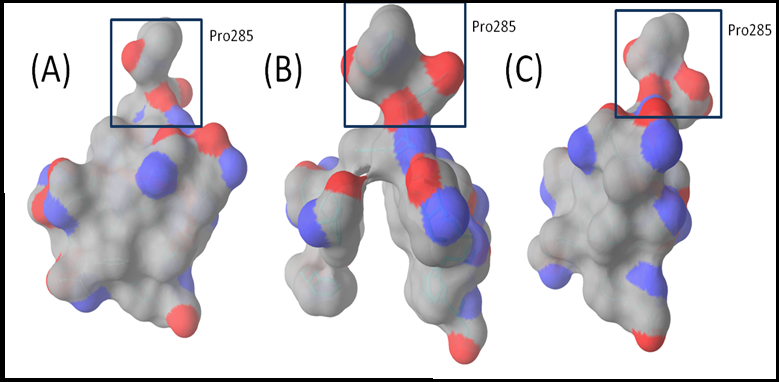
Figure 2: Binding Pocket :Top view of the binding pocket composed of F136, V139, F178, I277, A279, E280, 285, Y327, F610, V612, F615, F617, I626, and F628, shown as surface with carbons in grey of all three chains of AcrB, P285 which is situated in the back of the site is highlighted for reference. (A) The binding site in access protomer of 2J8S which is closed. (B) The binding site in binding protomer of 2J8S which is open. (C) The binding site in extrusion protomer of 2J8S which is collapsed after the substrate is extruded.
Docking of Known Substrates And Inhibitors
Native ligands of 2DRD and 2DR6 were docked in the binding site of their structures using IFD and QPLD and compared to the crystal complex in order to examine the reliability of the approach (Figure 1S). In case of minocycline, the binding takes place in the upper portion of the site away from the membrane surface which was classified as the ‘groove’ as it forms a narrow groove against Pro285; the poses were almost identical when compared to that of the co-crystal. In case of doxorubic in the ligand drifts towards the lower and wider portion of the site which was classified as cave, but can still be classified as mixed binder. This discrepancy can be attributed to the low resolution of 2DR6 (3.30Å). Since the binding mode of only two substrates are known, 14 substrates and two inhibitors (Figure 2S) were docked in the binding protomer of 2J8S to find out their binding modes in AcrB.
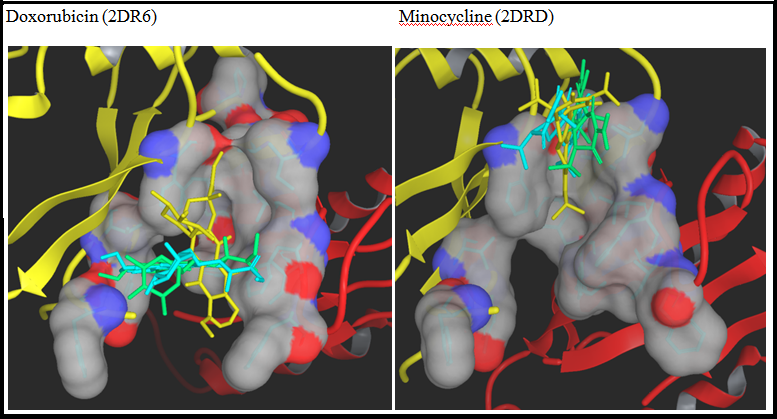
Figure 1s: Binding Modes of DOX and MIN: Superimposed binding modes of doxorubicin and minocycline in the substrate recognition site (side view of the binding pocket shown as a surface with carbon in grey, oxygen red and nitrogen blue) of binding protomer of AcrB in PDBID 2DR6 and 2DRD , respectively. PN2 and PC1 sub-domains of porter domain are shown in yellow and red cartoon representation, respectively. Ligands are shown in tube drawing with native, IFD and QPLD ligands in yellow, Light blue and spring green colors.
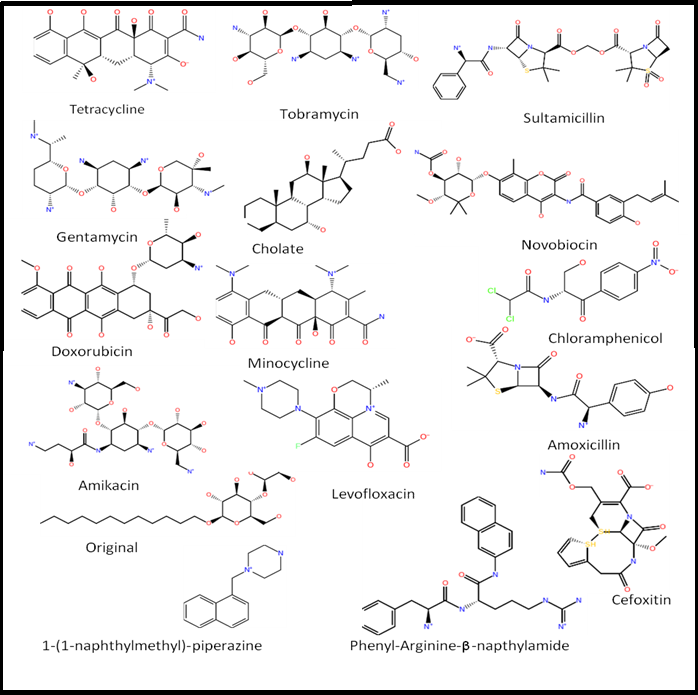
Figure 2s: Known Substrates :Two dimensional structures of known substrates of AcrB. Two inhibitors, NMP and PAβN are also shown.
Cave binding was exhibited by most of the substrates such as amikacin, amoxicillin, chloramphenicol, cholate, gentamycin, tetracycline, including doxorubicin and minocycline (Figure 3S). This is obvious as the binding site in 2J8S is narrower in comparison to the site in 2DR6 and 2DRD. Cefoxitin exhibits groove binding in IFD pose whereas levofloxacin, novobiocin, original, sultamicillin and tobramycin exhibit mixed binding pose in QPLD (Figure 4S). In terms of hydrogen bonding interactions, the substrates in IFD and QPLD seem to favor polar and aromatic residues such as Ser134, Ile 671, Glu673 and Gln176 (Table 1S & 2S). As the site is rich in phenylalanine, hydrophobic interactions also play a major role in stabilization of ligands in the active site through aromatic-aromatic interactions. In case of levofloxacin, it forms only one hydrogen bond with Phe136 in IFD whereas the QPLD pose is completely stabilized through aromatic-aromatic interactions. NMP and PAβN, the inhibitors of AcrB were found to exhibit cave and groove binding, respectively. Hydrogen bonding again suggests interactions of NMP and PAβN with polar residues in the site.

Figure 3s: IFD Binding Modes: IFD binding modes of known substrates and inhibitors in the substrate recognition site (side view of the binding pocket shown as a surface with carbon in grey, oxygen red and nitrogen blue) of binding protomer of AcrB. PN2 and PC1 sub-domains of porter domain are shown in yellow and red cartoon representation, respectively. Ligands are shown in tube drawing with green colored carbon atoms.

Figure 4s: QPLD Binding Modes: QPLD binding modes of known substrates and inhibitors in the substrate recognition site (side view of the binding pocket shown as a surface with carbon in grey, oxygen red and nitrogen blue) of binding protomer of AcrB. PN2 and PC1 sub-domains of porter domain are shown in yellow and red cartoon representation, respectively. Ligands are shown in tube drawing with green colored carbon atoms.
Table 1s: Molecular interactions of substrates and known inhibitors in the binding site as shown by Induced Fit Docking
| Molecules | Hydrogen bond donor | Hydrogen bond acceptor | Bond Length (Å) | Glide Docking Score | Glide Energy | IFD Score |
| Amikacin | LIG: N2 | SER134: O | 2.809 | -10.07 | -80.67 | -1809.58 |
| LIG: N3 | PRO 326: O | 2.921 | ||||
| LIG: N4 | TYR 327: OH | 3.011 | ||||
| LIG: N5 | GLN 176: O | 2.640 | ||||
| LIG: N5 | GLN 176: OE1 | 2.958 | ||||
| LIG: O10 | ILE 671: O | 2.850 | ||||
| LIG: O8 | PRO 326: O | 2.976 | ||||
| Amoxicillin | LIG: N3 | SER 134: O | 2.844 | -7.63 | -47.18 | -1802.42 |
| LIG: N3 | GLU 673:OE2 | 2.756 | ||||
| Cefoxitin | GLN 176: NE2 | LIG: O4 | 2.734 | -9.49 | -54.39 | -1805.57 |
| GLN 176:NE2 | LIG: O6 | 3.462 | ||||
| GLY 179: N | LIG: O6 | 3.168 | ||||
| Chloramphenicol | LIG: O1 | SER 630: OG | 3.187 | -8.26 | -46.30 | -1795.60 |
| Cholate | SER 134: OG | LIG: O3 | 2.982 | -9.63 | -45.72 | -1797.94 |
| GLU 673: N | LIG: O4 | 2.862 | ||||
| LIG: O3 | GLU 673: OE2 | 2.634 | ||||
| Doxorubicin | LIG: N1 | SER 134: OG | 2.909 | -10.50 | -64.67 | -1801.31 |
| LIG: N1 | GLU 673: OE2 | 2.668 | ||||
| LIG: O11 | GLU 673: OE2 | 2.715 | ||||
| LIG: O9 | OE2 ILE 671: O | 2.712 | ||||
| Gentamycin | LIG: N1 | SER 134: O | 2.915 | -10.85 | -66.47 | -1810.65 |
| LIG: N1 | ILE 671: O | 3.201 | ||||
| LIG: N3 | GLU 673: O | 2.732 | ||||
| LIG: O6 | ILE 671: O | 2.775 | ||||
| Levofloxacin | PHE 136: N | LIG: O3 | 2.708 | -9.86 | -37.04 | -1799.89 |
| Minocycline | GLU 673: N | LIG: O5 | 2.680 | -11.25 | -64.44 | -1798.75 |
| LIG: N2 | PRO 669: O | 3.227 | ||||
| LIG: N2 | ILE 671: O | 2.982 | ||||
| LIG: O1 | VAL 629: O | 2.935 | ||||
| Novobiocin | PHE 136: N | LIG: O3 | 3.179 | -12.67 | -76.26 | -1807.48 |
| PHE 136: N | LIG: O | 3.231 | ||||
| LIG: N2 | SER 134: O | 2.908 | ||||
| LIG: N2 | GLU 673: OE2 | 2.668 | ||||
| LIG: O11 | ALA 286: O | 2.962 | ||||
| LIG: O3 | SER 134: O | 2.804 | ||||
| Original | PHE 136: N | LIG: O4 | 3.334 | -9.44 | -61.61 | -1801.03 |
| LIG: O3 | TYR 327: OH | 2.731 | ||||
| LIG: O4 | SER 134: O | 3.125 | ||||
| LIG: O6 | PRO 669: O | 2.894 | ||||
| Sultamicillin | SER 134: OG | LIG: O7 | 3.172 | -12.20 | -79.40 | -1813.44 |
| PHE 136: N | LIG: O8 | 2.940 | ||||
| GLU 673: N | LIG: O9 | 3.089 | ||||
| LIG: N4 | ILE 671: O | 2.651 | ||||
| LIG: N4 | GLU 673: O | 2.811 | ||||
| Tetracycline | SER 134: OG | LIG: O6 | 2.690 | -10.41 | -59.35 | -1804.71 |
| PHE 136: N | LIG: O7 | 3.278 | ||||
| LIG: N1 | GLU 673: OE2 | 3.165 | ||||
| LIG: O7 | SER 134: O | 2.895 | ||||
| Tobramycin | GLN 89: NE2 | LIG: O3 | 2.920 | -7.63 | -97.15 | -1805.68 |
| LIG: N1 | PHE 617: O | 2.855 | ||||
| LIG: N1 | GLU 673: OE1 | 2.788 | ||||
| LIG: N1 | GLU 673: OE2 | 2.846 | ||||
| LIG: N2 | ASN 719: OD1 | 3.041 | ||||
| LIG: N2 | GLU 826: OE1 | 2.958 | ||||
| LIG: N3 | SER 80: O | 2.660 | ||||
| LIG: N3 | ASN 81: OD1 | 2.994 | ||||
| LIG: N3 | GLN 89: OE1 | 2.901 | ||||
| LIG: N4 | GLY 616: O | 3.012 | ||||
| LIG: N5 | ASP 681: OD2 | 2.758 | ||||
| LIG: N5 | GLU 826: OE2 | 2.670 | ||||
| LIG: O8 | GLU 683: OE1 | 2.602 | ||||
| ARG 815: NH1 | LIG: O7 | 3.363 | ||||
| ARG 815: NH2 | 2.843 | |||||
| NMP | LIG: N2 | VAL 629: O | 3.016 | -8.78 | -66.35 | -1792.29 |
| PAβN | LIG: N4 | PHE 178: O | 2.987 | -12.14 | -66.35 | -1809.10 |
| LIG: N5 | GLU 152: OE2 | 2.783 | ||||
| LIG: N5 | ILE 277: O | 2.952 | ||||
| LIG: N6 | GLU 152: OE1 | 2.579 | ||||
| LIG: N6 | SER 155: OG | 2.794 |
Table 2s: Molecular interactions of substrates and known inhibitors in the binding site as shown by Quantum-Polarized Ligand Docking
| Molecules | Hydrogen bond donor | Hydrogen bond acceptor | Bond Length (Å) | Glide Docking Score | Glide Energy |
| Amikacin | GLU 673: N | LIG: O12 | 2.870 | -9.05 | -84.33 |
| LIG: N3 | TYR 327: OH | 2.825 | |||
| LIG: N5 | GLU 673: OE2 | 2.632 | |||
| LIG: O12 | ILE 671: O | 3.523 | |||
| LIG: O13 | SER 134: OG | 2.819 | |||
| LIG: O8 | ILE 671: O | 2.643 | |||
| Amoxicillin | GLN 176: NE2 | LIG: O1 | 3.116 | -6.74 | -61.10 |
| LIG: N3 | SER 134: O | 2.795 | |||
| LIG: O5 | GLU 673: OE2 | 2.514 | |||
| Cefoxitin | PHE 136: N | LIG: O7 | 2.726 | -9.78 | -56.01 |
| LIG: N3 | SER 134: O | 2.655 | |||
| Chloramphenicol | PHE 136: N | LIG: O1 | 3.162 | -6.66 | -47.45 |
| LIG: N1 | SER 134: O | 2.978 | |||
| LIG: O1 | SER 134: O | 2.756 | |||
| Cholate | SER 135: OG | LIG: O4 | 3.026 | -10.56 | -54.79 |
| LIG: O3 | ILE 671: O | 2.515 | |||
| Doxorubicin | PHE 136: N | LIG: O9 | 2.167 | -14.49 | -70.96 |
| LIG: N1 | SER 134: OG | 2.734 | |||
| LIG: O11 | SER 134: OG | 2.815 | |||
| Gentamycin | ASN 719: ND2 | LIG: O6 | 3.109 | -8.75 | -113.23 |
| LIG: N3 | GLU 826: OE1 | 2.466 | |||
| LIG: N3 | GLU 826: OE2 | 3.158 | |||
| LIG: N4 | ASN 81: OD1 | 2.415 | |||
| LIG: N5 | ASP 83: OD1 | 2.722 | |||
| LIG: N5 | THR 87: OG1 | 2.630 | |||
| LIG: O7 | GLU 826: OE2 | 2.682 | |||
| Levofloxacin | -7.36 | -35.91 | |||
| Minocycline | GLU 673: N | LIG: O5 | 2.576 | -6.99 | -53.47 |
| LIG: N2 | GLU 673: OE2 | 3.097 | |||
| Novobiocin | LIG: N2 | GLY 179: O | 2.866 | -11.83 | -69.40 |
| LIG: O3 | LEU 177: O | 2.742 | |||
| Original | LIG: O3 | ILE 671: O | 2.573 | -12.48 | -61.66 |
| LIG: O4 | SER 134: O | 2.648 | |||
| LIG: O6 | GLU 673: OE2 | 3.217 | |||
| Sultamicillin | GLN 176: NE2 | LIG: O3 | 2.940 | -9.91 | -65.98 |
| LIG: N4 | ILE 671: O | 2.567 | |||
| Tetracycline | PHE 136: N | LIG: O6 | 2.757 | -9.02 | -60.76 |
| Tobramycin | SER 128: OG | LIG: O8 | 3.302 | -5.38 | -133.38 |
| LIG: N2 | GLU 130: OE1 | 2.405 | |||
| LIG: N2 | SER 134: OG | 2.973 | |||
| LIG: N2 | GLU 130: OE2 | 2.338 | |||
| LIG: N2 | GLN 176: OE1 | 3.333 | |||
| LIG: O3 | GLN 176: OE1 | 2.899 | |||
| LIG: O7 | SER 134: OG | 2.729 | |||
| LIG: O8 | GLU 130: OE1 | 2.872 | |||
| NMP | LIG: N2 | SER 134: OG | 2.516 | -9.34 | -44.09 |
| PAβN | LIG: N5 | GLU 130: OE1 | 2.572 | -9.91 | -73.95 |
| LIG: N6 | GLU 130: OE1 | 3.327 | |||
| LIG: N6 | GLU 130: OE2 | 2.486 |
Virtual Screening for Novel Leads
Five in silico small molecule libraries were computationally screened for interactions with the substrate recognition site leading to six potential hits. Firstly, PAβN was modified in which its aromatic scaffolds were replaced with aliphatic chains and these were screened against Pubchem for similar compounds. The resultant compounds were screened for interactions and PAβN_21341613 (Figure 3). Docking simulations of PAβN_21341613 suggested cave binding and it favors hydrogen bonding interactions with Gln569, Gln34, Tyr327, Ile 671 and Ser134 in both IFD and QPLD (Figure 4) (Table 1&2) which are polar and aromatic in nature. Secondly, Pubchem database was also screened for compounds similar to NMP, the resultant molecules were screened for interactions leading to two potential hits NMP_9993877 and NMP_10157779. Docking simulations of NMP_9993877 and NMP_10157779 suggested cave binding except for QPLD of NMP_10157779 which indicated mixed binding. Both the compounds favored hydrogen bonding interactions with Ser134, Ile671 and Glu673.
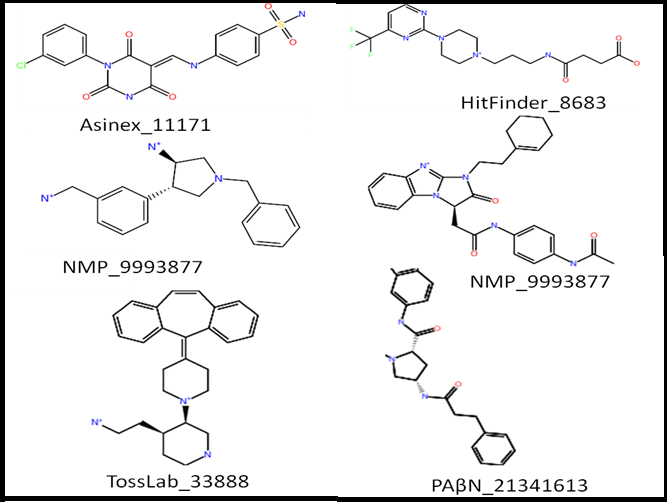
Figure 3: Lead Compounds :Two dimensional structures of Asinex_11171, HitFinder_8683, NMP_9993877, NMP_10157779, TOSLab_33888 and PAβN_21341613.
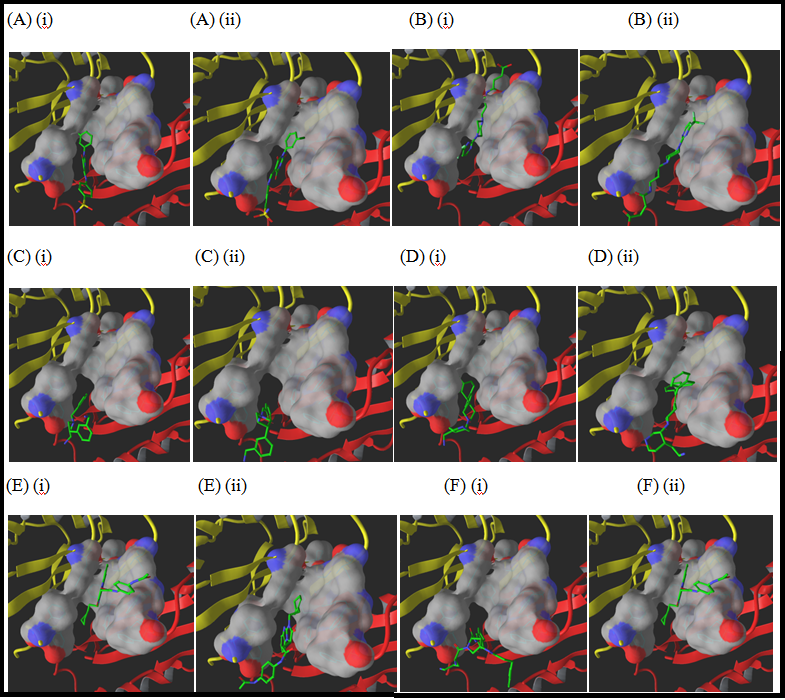
Figure 4: Binding Modes: IFD and QPLD binding modes of (A) Asinex_11171, (B) HitFinder_8683, (C) NMP_9993877, (D) NMP_10157779, (E) TOSLab_33888 and (F) PAβN_21341613 in the substrate recognition site (side view of the binding pocket shown as a surface with carbon in grey, oxygen red and nitrogen blue) of binding protomer of AcrB. PN2 and PC1 sub-domains of porter domain are shown in yellow and red cartoon representation, respectively. Ligands are shown in tube drawing with green colored carbon atoms. (i) and (ii) represents IFD and QPLD binding modes, respectively
Table 1: Molecular interactions of six potential hits in the binding site as shown by Induced Fit Docking
| Molecules | Hydrogen bond donor | Hydrogen bond acceptor | Bond Length (Å) | Glide Docking Score | Glide Energy | IFD Score |
| Asinex_11171 | LIG: N1 | PRO 669: O | 2.930 | -11.00 | -56.27 | -1800.19 |
| LIG: N1 | ILE 671: O | 2.789 | ||||
| Hitfinder_8683 | ALA 279: N | LIG: O1 | 2.903 | -10.81 | -57.08 | -1811.38 |
| LIG: N5 | PHE 178: O | 2.719 | ||||
| LIG: O3 | ASN 274: O | 2.667 | ||||
| NMP_99938777 | LIG: N2 | GLN 569: OE1 | 2.925 | -9.84 | -60.55 | -1803.87 |
| LIG: N2 | GLN 569: OE1 | 2..837 | ||||
| LIG: N2 | ILE 671: O | 2.970 | ||||
| LIG: N3 | SER 134: O | 3.380 | ||||
| LIG: N3 | GLU 673: OE2 | 2.586 | ||||
| NMP_10157779 | LIG: N2 | GLU 673: OE2 | 2.726 | -12.07 | -61.98 | -1804.03 |
| LIG: N3 | PRO 669: O | 2.735 | ||||
| LIG: N3 | ALA 670: O | 3.095 | ||||
| LIG: N3 | ILE 671: O | 2.657 | ||||
| TossLab_33888 | ARG 620: NE | LIG: O3 | 2.851 | -11.42 | -57.40 | -1803.48 |
| LIG: N5 | GLN 89: OE1 | 2.769 | ||||
| PAβN_213413 | PHE 136: N | LIG: O1 | 3.522 | -12.07 | -78.08 | -1807.87 |
| LIG: N2 | GLU 673: OE1 | 3.025 | ||||
| LIG: N4 | SER 134: O | 2.889 | ||||
| LIG: N4 | ILE 671: O | 3.005 | ||||
| LIG: N5 | GLN 34: OE1 | 2.731 | ||||
| LIG: N5 | TYR 327: OH | 2.946 | ||||
| LIG: N5 | GLN 569: OE1 | 2.841 |
Table 2: Molecular interactions of six potential hits in the binding site as shown by Quantum-Polarized Ligand Docking
| Molecules | Hydrogen bond donor | Hydrogen bond acceptor | Bond Length (Å) | Glide Docking Score | Glide Energy |
| Asinex_11171 | LIG: N1 | SER 134: O | 2.729 | -9.99 | -60.56 |
| HitFinder_8683 | SER 135: OG | LIG: O2 | 2.676 | -11.17 | -73.58 |
| PHE 136: N | LIG: O1 | 2.829 | |||
| LIG: O3 | ILE 671: O | 2.679 | |||
| NMP_9993877 | LIG: N2 | SER 134: O | 2.575 | -9.11 | -76.30 |
| LIG: N2 | SER 134: OG | 2.290 | |||
| LIG: N3 | PRO 669: O | 2.680 | |||
| LIG: N3 | ILE 671: O | 2.523 | |||
| NMP_10157779 | LIG: N2 | SER 134: O | 2.761 | -9.58 | -62.69 |
| LIG: N3 | GLU 673: OE2 | 2.630 | |||
| TossLab_33888 | SER 135: OG | LIG: O3 | 2.677 | -8.50 | -66.71 |
Three freely available subsets of commercial small molecule databases- Maybridge HitFinder, Asinex and TOSLab were also screened which generated one potential hit each. Maybridge HitFinder generated Hitfinder_8683 which exhibited groove and mixed binding in IFD and QPLD, respectively. Asinex Database generated Asinex_11171 which exhibited cave binding in both IFD and QPLD. TOSLab Database generated TOSLab_33888 which exhibited mixed binding in both IFD and QPLD. These compounds favored hydrogen bonding interactions with mainly polar and aromatic residues such as Ser134, Phe178 and Ile671. The docking modes of all the above ligands were superimposed on the 3D - SiteMap of the binding site to find out any clashes (such as donor group buried in an acceptor region). Such maps are particularly important for lead optimization as the moieties present in the regions which are neither hydrophobic nor hydrophilic can be useful for further substitution to improve the physical properties of the ligand with minimal effect on binding affinity. Five out of six ligands follow the Lipinski rule-of-five which to assess whether a compound is drug-like (Table 3S). PAβN_21341613 has six H- donors but it was still selected as it is a derivative of PAβN which has 7.5 H-donors. The glide emodel and glide energy of most of the compounds are very good with relatively less hydrogen bonds which suggests that aromatic-aromatic interactions also play a major role in stabilizing the ligands in the binding site. When administered along with an antibiotic, these compounds can increase intracellular antibiotic concentrations and thereby restoring activity of the antibiotics and hence they can emerge as potent inhibitors of multidrug efflux pump AcrB.
Table 3s: Lipinski properties of the docked ligands as calculated by Qikprop 3.2
| Molecules | Molecular weight | Log P (o/w) | H-donor | H-acceptor |
| Asinex_11171 | 420.826 | 1.194 | 3 | 8.5 |
| HitFinder_8683 | 389.377 | -0.073 | 2 | 9 |
| NMP_9993877 | 281.4 | 1.241 | 4 | 4 |
| NMP_10157779 | 399.578 | 3.403 | 3 | 4.5 |
| TOSLab_33888 | 471.558 | 3.762 | 2 | 9.5 |
| PAβN_21341613 | 491.632 | 1.245 | 6 | 10 |
id="CONCLUSION"Conclusion
The selected ligands share most crucial residues with that of known substrates and inhibitors along with the binding modes. The aromatic components are responsible for better aromatic- aromatic interactions with the binding site. These inhibitors can form more powerful interactions with the binding site compared to substrates and arrest the conformational change in AcrB. Nearly all the ligands reported here provide this possibility of further elaboration and optimization because of their lead like properties, relatively small size, well balanced physico-chemical properties and the absence of chemical functionalities associated with metabolic or toxic liabilities.
Acknowledgment
The authors thank DBT-AIST (Japan) for financial support for this research and to purchase Schrödinger software. DV thanks DST-FIST and UGC-SAP for funding facilities to the Centre for Advanced Study in Crystallography and Biophysics. Bioinformatics Infrastructure Facility provided to the University of Madras by the Department of Biotechnology, India is gratefully acknowledged. There is no conflict of interest.
References
- 1. Levy, S. B., Marshall, B. Antibacterial resistance worldwide: causes, challenges and responses. (2004) Nat Med 10(12): S122- S129.
- 2. Bryan, L.E., Bedard, J., Wong, S., et al. Quinolone antimicrobial agents: mechanism of action and resistance development. (1989) Clin Invest Med 12(1): 14- 19.
- 3. Speer, B.S., Shoemaker, N.B., Salyers, A.A. Bacterial resistance to tetracycline, mechanisms, transfer, and clinical significance. (1992) Clin Microbiol Rev 5(4): 387- 399.
- 4. Ma, D., Cook, D.N., Hearst, J.E., et al. Efflux pumps and drug resistance in gram negative bacteria. (1994) Trends Microbiol 2(12): 489- 493.
- 5. Marshall, N.J., Piddock, N.J. Antibacterial efflux systems. (1997) Microbiologia 13(3): 285- 300.
- 6. Nikaido, H. Multidrug efflux pumps of gram-negative bacteria. (1996) J Bacteriol 178(20): 5853- 5859.
- 7. Stratton, C. W. Mechanisms of bacterial resistance to antimicrobial agents. (2000) J Med Liban 48(4): 186-198.
- 8. Wright, G. D. Aminoglycoside-modifying enzymes. (1999) Curr Opin Microbiol 2(5): 499- 503.
- 9. Banerjee, S.K., Bhatt, K., Misra, P., et al. Involvement of a natural transport system in the process of efflux- mediated drug resistance in Mycobacterium smegmatis. (2000) Mol Gen Genet 262(6): 949- 956.
- 10. Lee, V.J., Hecker, S.J. Antibiotic resistance versus small molecules, the chemical evolution. (1999) Med Res Rev 19(6): 521- 542.
- 11. Li, X.Z., Zhang, L., Poole, K. Role of the multidrug efflux systems of Pseudomonas aeruginosa in organic solvent tolerance. (1998) J Bacteriol 180(11): 2987- 2991.
- 12. Kobayashi, N., Nishino, K., Yamaguchi, A. Novel macrolide-specific ABC-type efflux transporter in Escherischia coli. (2001) J Bacteriol 183(19): 5639- 5644.
- 13. Nikaido, H., Basina, M., Nguyen, V., et al. Multidrug efflux pump AcrAB of Salmonella typhimurium excretes only those beta-lactum antibiotics containing lipophilic side chains. (1998) J Bacteriol 180(17): 4686-4692.
- 14. Levy, S.B., McMurry, L. Plasmid-determined tetracycline resistance involves new transport systems for tetracycline. (1978) Nature 276(5683): 90- 92.
- 15. McMurry, L., Petrucci, R.E., Levy, S.B. Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. (1980) Proc Natl Acad Sci USA 77(7): 3974- 3977.
- 16. Ball, P.R., Shales, S.W., Chopra, I. Plasmid-mediated tetracycline resistance in Escherichia coli involves increased efflux of the antibiotic. (1980) Biochem Biophys Res Commun 93(1): 74- 81.
- 17. Hirakata, Y., Srikumar, R., Poole, K., et al. Multidrug efflux systems play an important role in the invasiveness of Pseudomonas aeruginosa. (2002) J Exp Med 196(1): 109- 118.
- 18. Van Dyk, T.K., Templeton, L.J., Cantera, K.A., et al. Characterization of the Escherichia coli AeaAB efflux pump: metabolic relief valve? (2004) J Bacterial 186(21): 7196- 7204.
- 19. Poole, K. Mechanisms of bacterial biocide and antibiotic resistance. (2002) J Appl Microbiol 92: 55S- 64S.
- 20. Thanassi, D.G., Cheng, L.W., Nikaido, H. Active efflux of bile salts by Escherischia coli. (1997) J Bacteriol 179(8): 2512- 2518.
- 21. Sulavik, M.C., Houseweart, C., Cramer, C., et al. Antibiotic Susceptibility Profiles of Escherichia coli Strains Lacking Multidrug Efflux Pump Genes. (2001) Antimicrob Agents Chemother 45(4): 1126– 1136.
- 22. Zgurskaya, H.I., Nikaido, H. Bypassing the periplasm: Reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. (1999) Proc Natl Acad Sci 96(13): 7190- 7195.
- 23. Dinh, T., Paulsen, I.T., Saier, M.H. A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. (1994) J Bacteriol 176(13): 3825– 3831.
- 24. Murakami, S., Nakashima, R., Yamashita, E., et al. Crystal structure of bacterial multidrug efflux transporter AcrB. (2002) Nature 419(6907): 587- 593.
- 25. Murakami, S., Nakashima, R., Yamashita, E., et al. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. (2006) Nature 443(7108): 173- 179.
- 26. Seeger, M.A., Diederichs, K., Eicher, T. et al. The AcrB Efflux Pump: Conformational Cycling and Peristalsis Lead to Multidrug Resistance. (2008) Current Drug Targets 9(9): 729- 749.
- 27. Sennhauser, G., Amstutz, P., Briand, C., et al. Drug Export Pathway of Multidrug Exporter AcrB Revealed by DARPin Inhibitors. (2007) PLoS Biol 5: 106- 113.
- 28. Boyer, P. D. The ATP synthase - a splendid molecular machine. (1997) Annu Rev Biochem 66: 717- 749.
- 29. Yao, X.Q., Kenzaki, H., Murakami, S., et al. Drug export and allosteric coupling in a multidrug transporter revealed by molecular simulations. (2010) Nature Comm 1: 117.
- 30. Schulz, R., Vargiu, A.V., Collu, F., et al. Funtional rotation of the transporter AcrB: Insights into Drug Extrusion from simulations. (2010) PLos Comput Biol 6(6): 1- 10.
- 31. Feng, Z., Hou, T., Li, Y. Unidirectional peristaltic movement in multisite drug binding pockets of AcrB from molecular dynamics simulations. (2012) Mol BioSyst 8(10): 2699- 2709.
- 32. Mahamoud, A., Chevalier, J., Alibert-Franco, S., et al. Antibiotic efflux pumps in Gram-negative bacteria: the inhibitor response strategy. (2007) J Antimicrob Chemother 59(6): 1223- 1229.
- 33. Piddock, L.J., Whale, K., Wise, R. Quinolone resistance in Salmonella: clinical experience. (1990) Lancet 335(8703): 1459.
- 34. Lewis, K. In search of natural substrates and inhibitors of MDR pumps. (2001) J Mol Microbiol Biotechnol 3(2): 247– 254.
- 35. Kaatz, G.W. Bacterial efflux pump inhibition. (2005) Curr Opin Investig Drugs 6(2): 191– 198
- 36. Lomovskaya, O., Bostian, K.A. Practical applications and feasibility of efflux pump inhibitors in the clinic—a vision for applied use. (2006) Biochem Pharmacol 71(7): 910– 918.
- 37. Schumacher, A., Steinke, P., Bohnert, J.A., et al. Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Enterobacteriaceae other than Escherichia coli. (2006) J Antimicrob Chemother 57(2): 344– 348.
- 38. Takatsuka, Y., Chen, C., Nikaido, H. Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. (2010) Proc Natl Acad Sci 107(15): 6559- 6565.
- 39. Yadav, P.K., Singh, G., Gautam, B., et al. Molecular modeling, dynamics studies and virtual screening of Fructose 1,6 biphosphate aldolase-II in community acquired- methicillin resistant Staphylococcus aureus (CA-MRSA). (2013) Bioinformation 9(3): 158- 164.
- 40. Farmer, R., Gautam, B., Singh, S., et al. Virtual screening of AmpC / β – Lactamase for antimicrobial resistance in Pseudomonas aeruginosa. (2010) Bioinformation 4(7): 290- 294.
- 41. Bolton, E., Wang, Y., Thiessen, P.A., et al. PubChem: Integrated Platform of Small Molecules and Biological Activities. (2008) Annu Rep Comput Chem 4: 217- 241.
- 42. Shelley, J.C., Cholleti, A., Frye, L.L., et al. Epik: a software program for pK (a) prediction and protonation state generation for drug-like molecules. (2007) J Comput Aided Mol Des 21(12): 681- 691.
- 43. Jorgensen, W.L., Maxwell, D.S., Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. (1996) J Amer Chem Soc 118(45): 11225- 11236.
- 44. Sherman, W., Day, T., Jacobson, M.P., et al. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. (2006) J Med Chem 49(2): 534- 553.
- 45. Cho, A.E., Guallar, V., Berne, B., et al. Importance of Accurate Charges in Molecular Docking: Quantum mechanical/molecular mechanical (QM/MM) Approach. (2005) J Comput Chem 26(9): 915- 931.
- 46. Nelson, M. L. Modulation of Antibiotic Efflux in Bacteria. (2002) Curr Med Chem Anti-Infective Agents 1 :35- 54.
- 47. Pos, K.M. Trinity revealed: Stoichiometric complex assembly of a bacterial multidrug efflux pump. (2009) Proc Natl Acad Sci 106(17): 6893- 6894.










