
Targeting Monoclonal Antibodies to the Tumor Microenvironment for Cancer Therapy/Immunotherapy
Frederick L. Hall1, Sant P. Chawla2, Neal S. Chawla2, Erlinda M. Gordon1,2*
Affiliation
- 1Counterpoint Biomedica LLC, Santa Monica CA 90403, USA
- 2Sarcoma Oncology Center/Cancer Center of Southern California, Santa Monica CA 90403, USA
Corresponding Author
Erlinda M. Gordon, 2811 Wilshire Blvd., Suite 414, Santa Monica CA 90403, USA; Tel: 818-726-3278; E-mail: erlinda.gordon@gmail.com
Citation
Gordon, EM., et al. Targeting Monoclonal Antibodies to the Tumor Microenvironment for Cancer Therapy / Immunotherapy (2016) Cell Immunol Serum Biol 2(1): 20- 26.
Copy rights
Copy rights: ©2016 Gordon, EM. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Monoclonal Antibodies, Immune Checkpoint Inhibitors, Tumor- Targeting, Targeted Drug Delivery, Tumor Microenvironment, Extracellular Matrix
Abstract
Background: Therapeutic antibodies and immune checkpoint inhibitors can be delivered more efficiently to the tumor microenvironment (TME) by targeting the exposed collagenous (XC) proteins at the site[s] of tumor invasion, stroma formation, and neoangiogenesis.
Purpose: To assess the ability of bifunctional fusion polypepetides (mAb-Tropins) to selectively deliver monoclonal antibodies (mAbs) to the TME in tumor-bearing nude mice.
Methods: Synthetic peptide probes and polypeptide aptamers (25- 50 aa), fluorescein -labeled IgGs, and an anti-VEGF human mAb were tested in vitro in exposed collagen (XC)-agarose binding assays, in HUVEC cultures, and in vivo in a human xenograft model of pancreatic cancer in nude mice.
Results: The XC-targeted mAb-Tropins, bound non-covalently to FITC-labeled IgGs [but not to truncated F(Ab’)2 fragments], and exhibited selectivity for XC-agarose over control agarose matrices. Importantly, the biological activity of the XC/mAb-Tropinbound anti-VEGF mAbs was fully preserved, as demonstrated by inhibition of endothelial cell proliferation in HUVEC cultures. In vivo, intense fluorescence was observed in tumors of mice injected with mAb-Tropin targeted IgGs at 15 and 60 minutes after intravenous injection, but not in non-targeted IgG-treated mice.
Conclusions: Tumor XC-targeted mAb-Tropins are an effective method of delivering therapeutic mAbs precisely to the tumor compartments, with meaningful implications for cancer therapy/immunotherapy.
Introduction
Humanized monoclonal antibodies (mAbs) are now well established as “targeted therapies” for a wide variety of malignancies[1], but there remains a host of problems with systemic biodistribution, target selectivity, poor tumor penetration, and untoward toxicities, which limit their clinical utility[2,3]. Likewise, the new generation of mAbs, designed to break immune tolerance and enhance immune responses by inhibiting T-cell checkpoints, offers another promising avenue of tumor control[4,5] combination regimens using immune checkpoint inhibitors have resulted in greater response rates than those achieved with standard chemotherapy for unrespectable or metastatic melanoma[6] but the ungoverned biodistribution of immune checkpoint inhibitors are associated with severe and/or serious immune-related adverse events (irAEs)[7-9], and a significant percentage of patients will exhibit primary resistance to these inhibitors[10,11].
Among the acquired cellular, genetic, and biochemical “Hallmarks of Cancer” that collectively dictate malignant cell growth[12], is the influential “tumor microenvironment” (TME) of stromal elements provided by resident fibroblasts, endothelial cells, pericytes, leukocytes, and extracellular matrix (ECM) components, which functions as an integral part of cancer initiation, growth, and progression, as well as tissue invasion and metastasis[13,14]. With growing appreciation of the TME as an essential histopathological component of cancer[15,16], advanced therapies targeting the cellular and non-cellular compartments of tumors are being designed and applied in the clinic[17]. In this article, we report on a new platform biotechnology and scientific approach to the pro-active targeting of specific proteinaceous components of the TME which could provide more selective and efficient mAb delivery to tumor compartments, and potentially enhancing mAb efficacy and reducing systemic toxicity, thus providing more favorable outcomes for cancer therapy/immunotherapy.
Materials and Methods
Synthetic peptide targeting probes ( < 40 aa) and polypeptide aptamers (40 - 50 aa), 85 - 99% purity, were prepared by 9-fluorenylmethyloxycarbonyl (Fmoc) solid phase peptide synthesis, purified by high performance liquid chromatography (HPLC), and verified by mass spectrometry and amino acid analysis. FITC-labeled probes and mAb-Tropins were synthesized by Biomatik; additionally, experimental mAb-Tropins were synthesized by C.S. Bio Co. and Thermo-Fisher with similar purities. Polypropylene mini-columns (2.5 ml), FITC labeled rabbit anti-human IgGs (whole molecule), F(Ab’)2 fragments, and collagen XC-agarose chromatography matrix (1 mg/ml collagen type III) were purchased from Sigma Aldrich. Anti-VEGF IgG1 (bevacizumab, Avastin, Genentech/Roche) was stored neat in aliquots (25 mg/ml stock) for cell culture experiments or was coupled to Oregon Green-488 for binding studies using an Alexa Fluor 488 protein labeling kit (Life Technologies) according to the manufacturer’s instructions. Analysis of fluorescence in XC-binding assays was visualized with an Ultra Bright Blue Light transilluminator equipped with an amber filter (New England Bio Group); photo-documentation was provided by a Leica V-Lux 1 digital camera; and fluorescence was quantified using a Quantus fluorometer (Promega).
Chromatographic Collagen XC-Agarose Binding Assays
For comparative XC-binding studies, linear polypeptide aptamers (5 mg) were suspended in 200 μl of 50% dimethyl sulfoxide (DMSO) in PBS, pH 7.4, followed by dilution first to 5 mg/ml (10% DMSO) stock solutions and then to 1 mg/ml (2% DMSO) working concentrations which were stored frozen at -20°C. Varying amounts of the mAb-tropin onco-aptamers were mixed with FITC-labeled IgGs or Oregon Green-labeled anti-VEGF monoclonal antibody in PBS, pH 7.4, at room temperature for 30 minutes, and the reaction mixtures were applied to the equilibrated mini-columns containing 250 ml bed volume of XC-agarose beads, followed by stringent washing (>10 column volumes) with PBS, PBST (PBS + 0.5% Tween-20), and 1.0 M NaCl in PBST, respectively. Following photo-documentation of the comparative fluorescence retained on the columns, both the initial column pass-through eluates and the re-suspended retentates were analyzed by fluorometry.
Human Umbilical Vein Endothelial Cells (HUVEC, Cat: C-003-5C), obtained from Cascade Biologics Gibco/Life Technologies), were maintained at 37°C and 5% CO2 in Gibco 200 Basal Medium (M200) supplemented with LVES (2% FBS, hydrocortisone, EGF, bFGF, heparin, and ascorbic acid), prior to experimentation in low serum 0.2% fetal bovine serum (FBS, 4 hours) followed by a spike in 0.2% FBS with 10 ng/ml recombinant human VEGF (Gibco/Life Technologies). Documentation of HUVEC cell growth in 48 well plates was obtained with an inverted microscope using phase contrast optics, followed by actual cell counts in the presence of Trypan Blue with a Bright Line Neubauer Hemocytometer. To verify that the biologic function of the anti-VEGF IgG1 was retained as a VEGF-Trap while bound by mAb-Tropins to XC-agarose beads, two sterile XC-agarose columns were prepared: reaction mixtures containing (i) Anti-VEGF IgG1 only or (ii) Anti-VEGF IgG1 plus mAb-Tropin were applied to the respective columns followed by extensive washing. (Note: a fluorescent anti-VEGF IgG1-Green tag was spiked into each reaction mixture to confirm washing stringency and aptamer-dependent anti-VEGF IgG1binding) Following HUVEC plating in 48 well plates and serum deprivation in 0.2% FBS for 4 hours, equal amounts of VEGF-spiked growth medium was passed over one of the two prepared test columns, and the resulting eluates were evaluated for the ability of the remaining VEGF to stimulate the proliferation of the serum-deprived endothelial cells. Representative photo-micrographs and resulting cell counts were obtained after 24 hours.
Tumor Targeting in a Mouse Model of Metastatic Cancer
The following animal studies were performed in collaboration with Anti Cancer, Inc., San Diego, California, under a protocol approved by the University of California San Diego, Institutional Animal Care and Use Committee, Protocol Number 15 - 092). Five to six-week old NCR nu/nu mice received a subcutaneous injection of 5x106 MiaPaca-2 human pancreatic cancer cells into the right flanks. When the average tumor xenograft reached a volume of approximately 50 mm³, test samples containing fluorescent tumor-targeting probes or fluorescent IgGs in the presence or absence of mAb-Tropin onco-aptamers were injected into the tail veins, followed by evaluation of tumor fluorescence over time using an Olympus OV 100 Fluorescent Imaging System (a closed-chamber system for high-sensitivity, high-speed imaging with minimal specimen damage). Following high-sensitivity analysis of the excised tumors, additional biopsies of liver, heart, skeletal muscle, colon, and bone were examined to assess biodistribution in non-target organs.
Results and Discussion
Based on the pathological exposure of collagenous proteins (XC-proteins) within the tumor microenvironment, the molecular engineering of a high affinity collagen XC-binding domain contained within the D2 propeptide of von Willebrand coagulation Factor (vWF)[18,19] which normally guides growth-factor laden platelets to injured and/or diseased tissues has been adapted to the design and development of an active tumor-targeting drug delivery platform (“Pathotropic Targeting”) that has been tested and proven to be effective in clinical trials[20-23]. This platform, which enabled the development of Rexin-G and Reximmune-C, the first, and so far only, targeted, injectable gene therapy vectors to be validated in the clinic[24,25], has been further engineered and incorporated into a new series of bifunctional adaptor polypeptides (called onco-aptamers, see Figure 1), which are designed to deliver chemotherapies and biologic therapies (Figure 1A), including therapeutic mAbs (Figure 1B), to tumors without altering either the chemical composition or the commercial manufacturing process of the drugs approved by the FDA. Leveraging the molecular histopathology of the TME via a high affinity XC-protein binding domain on the one hand, and a high-affinity immunoglobulin (IgG)-binding domain on the other hand, the resulting IgG-targeting onco-aptamers, named mAb-Tropins, are designed to enable the monoclonal antibodies to accumulate and enhance effective drug concentration in the TME, and improve anti-tumor efficacy while reducing systemic toxicity.
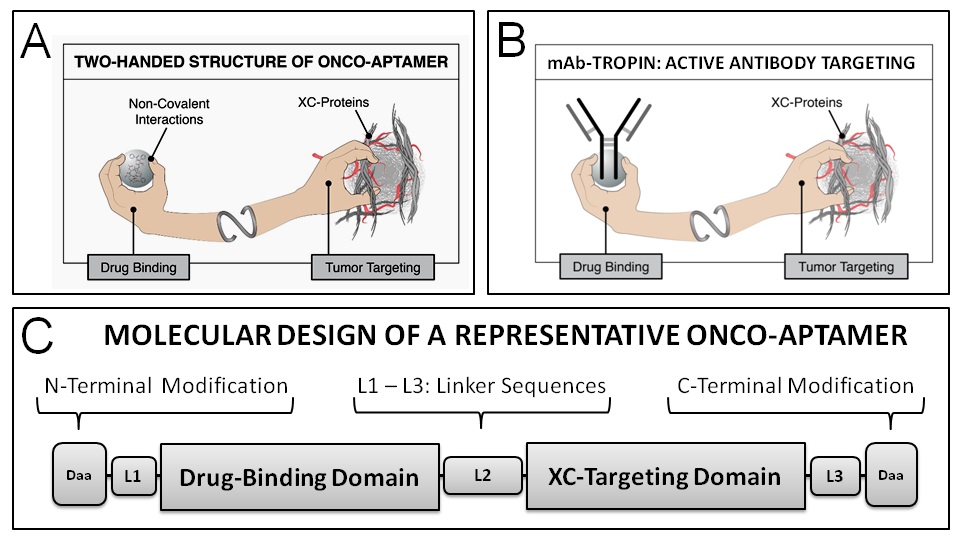
Figure 1: Core Design Elements of Onco-Aptamers. As shown in the schematic illustration, an onco-aptamer is a fusion polypeptide that contains two or more aptamer sequences: (i) a tumor-targeting domain containing sequences that bind to exposed collagenous (XC) proteins found abundantly in the tumor microenvironment, and (ii) a drug binding domain containing sequences that bind to specific classes of chemotherapeutic or biologic agents (A), including mAb-Tropins that are specific for IgG/mAbs (B). The drug-binding functionality of these fusion polypeptides is based on high-affinity, non-covalent interactions. N-terminal and C-terminal modifications (C) are designed to enhance stability and function in vivo.
A chromatographic simulation of histological tumor-targeting in vitro is shown in Figure 2. The XC-targeting mAb-Tropin, bound non-covalently to fluorescein isothiocyanate (FITC)-labeled IgGs (a fluorescent cargo), is shown to be selective for XC-agarose (a proxy for tumor tissue) over control agarose (normal tissue). Further, the localization of the mAb-Tropin binding site to the C-terminal constant Fc region of the IgG heavy chain was demonstrated using FITC-labeled whole IgGs (Figure 3A-1,2) in comparison with FITC-labeled F(Ab’)2 fragments (Figure 3A-3)-proteolytic fragments in which the Fc regions have been digested and truncated by pepsin treatment. XC-agarose chromatography (Figure 3B) shows mAb-Tropin dependent binding of whole fluorescent IgGs to the XC-agarose matrix (Figure 3B-2, Positive), but not F(Ab’)2 fragments (Figure 3B-3, Test, Negative), which contain the functional antigen-binding sites.
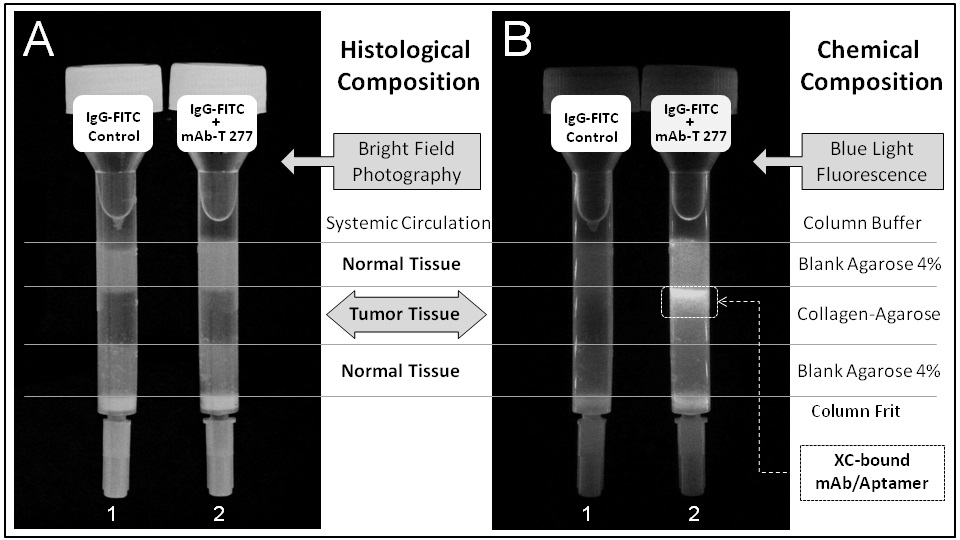
Figure 2: Chromatographic simulation of histological tumor-targeting in vitro. The XC-targeting mAb-Tropin 277, bound non-covalently to FITC-labeled IgGs (a fluorescent cargo), is shown to be selective for XC-agarose (a proxy for tumor tissue) over control agarose (normal tissue). Importantly, the biological activity of the XC/Aptamer-bound mAb is fully preserved, by design, since the IgG-binding site of the mAb-Tropin is located at the C-terminal constant Fc region of the heavy chain, far from the antigen-binding sites. A, Bright Field; B, Fluorescence.
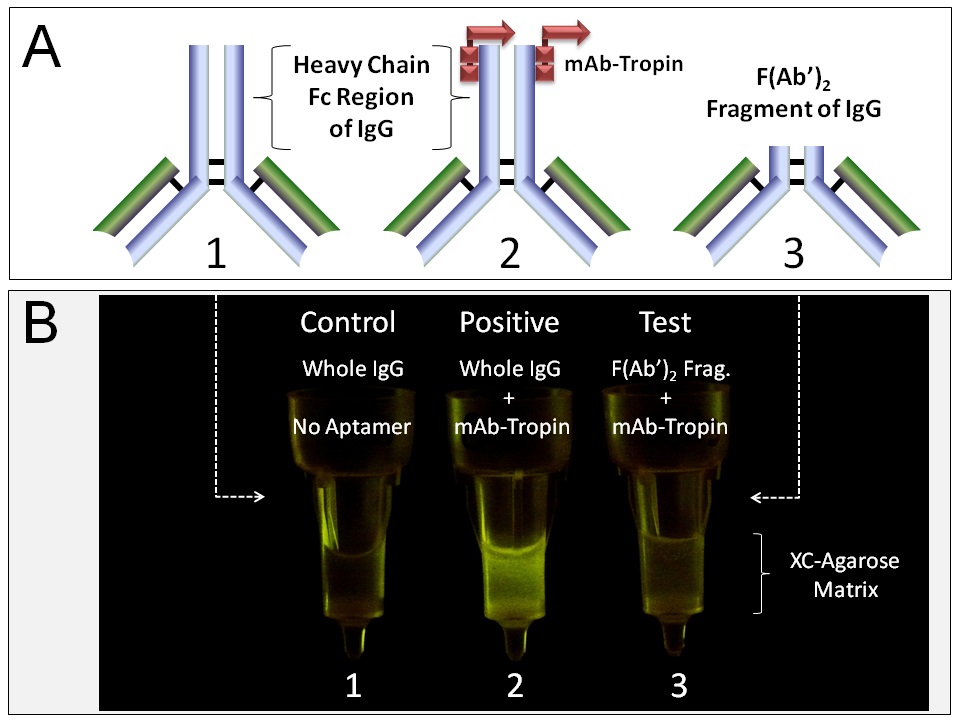
Figure 3: Differential binding of mAb-Tropins to whole IgGsvsF(Ab’)2 fragments. The localization of the mAb-Tropin binding site to the C-terminal constant Fc region of the IgG heavy chain was demonstrated using FITC-labeled whole IgGs (A 1, 2) in comparison with FITC-labeled F(Ab’)2 fragments (A 3)—proteolytic fragments in which the Fc regions have been digested and truncated by pepsin treatment. XC-agarose chromatography (B) shows mAb-Tropin dependent binding of whole fluorescent IgGs to the XC-agarose matrix (B 2, Positive), but not F(Ab’)2 fragments (B 3, Test, negative), which contain the functional antigen-binding sites.
Importantly, the biological activity of the XC/Aptamer-bound mAb is, by design, fully preserved: the IgG-binding site of the mAb-Tropin is located at the C-terminal constant Fc region of the IgG heavy chain (Figure 3), far from the antigen-binding sites. The conservation of biological activity of an XC/Aptamer/IgG complex was verified using a monoclonal antibody against vascular endothelial growth factor (VEGF, bevacizumab) and XC-agarose column chromatography, which together served as a VEGF-Trap to remove the growth factor from VEGF-supplemented cell culture medium, thus preventing the classic VEGF-induced stimulation of endothelial cell proliferation (Figure4).
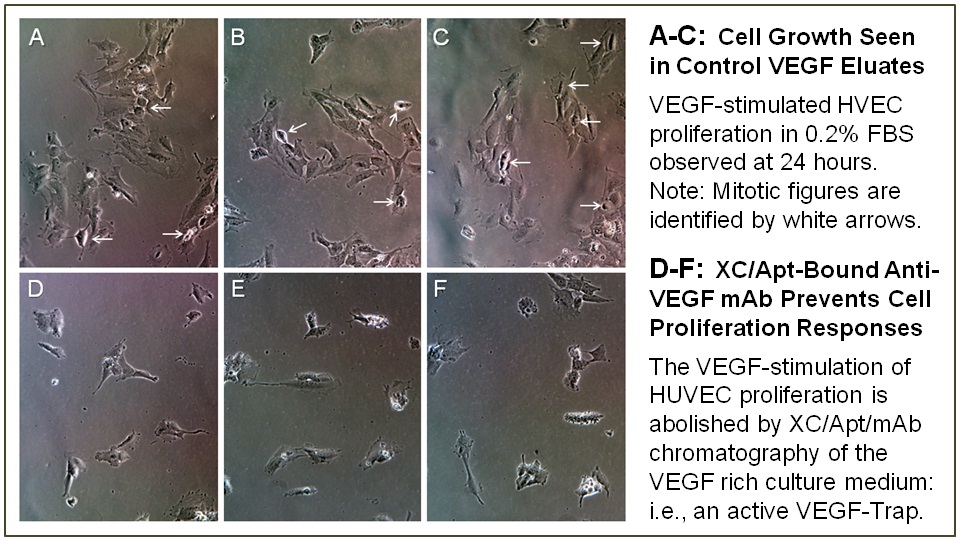
Figure 4: Demonstration that XC/ mAb-Tropin/ bevacizumab bound complexes retain anti-VEGF bioactivity. XC-column bound mAb-Tropin/bevacizumab complexes were used as a VEGF-Trap to deplete the VEGF growth factor from VEGF-supplemented culture medium, thereby preventing VEGF-dependent stimulation of Human Vascular Endothelial Cell (HVEC) proliferation, as is shown in cell culture. The representative upper plates (A-C), shown in triplicate, reveal mitotic figures and endothelial cell proliferation in HUVEC cultures stimulated with VEGF at 24 hours. In contrast, the lower plates (D-F), shown in triplicate, demonstrate that the VEGF-stimulation of HUVEC cells seen at 24 hours is prevented by passage of the VEGF-spiked medium through the XC/mAb-Tropin/bevacizumab XC-column—indicating that the bevacizumab bound non-covalently to the mAb-Tropin is biologically active.
As shown in Figure 5A-5B, the bifunctional mAb-Tropin 277 binds tightly to IgGs upon simple mixing, efficiently tethering the mAb-Tropin/IgG complexes to the XC-agarose matrices while quantitatively removing the IgGs from the “circulation”/solution. The physiological and pharmacological advantages of active XC(tumor)-targeting of therapeutic mAbs, over passive accumulation in tumors, were examined further in a murine xenograft model of metastatic pancreatic cancer using fluorescent IgGs to monitor biodistribution. As shown in Figure 5C, upon intravenous infusion of FITC-IgGs in the presence or absence of the mAb-tropin targeting aptamers, the tumors of IgG + mAb-Tropin 277 treated mice revealed rapid and detectable accumulations of fluorescent IgGs in tumors at 15 minutes and 60 minutes, while non-targeted IgGs failed to indicate significant accumulations beyond the background fluorescence seen in control non-treated animals. Fluorescence seen in the non-target organs of IgG + mAb-Tropin 277 treated mice was consistently less than that seen in non-targeted IgG treated animals.
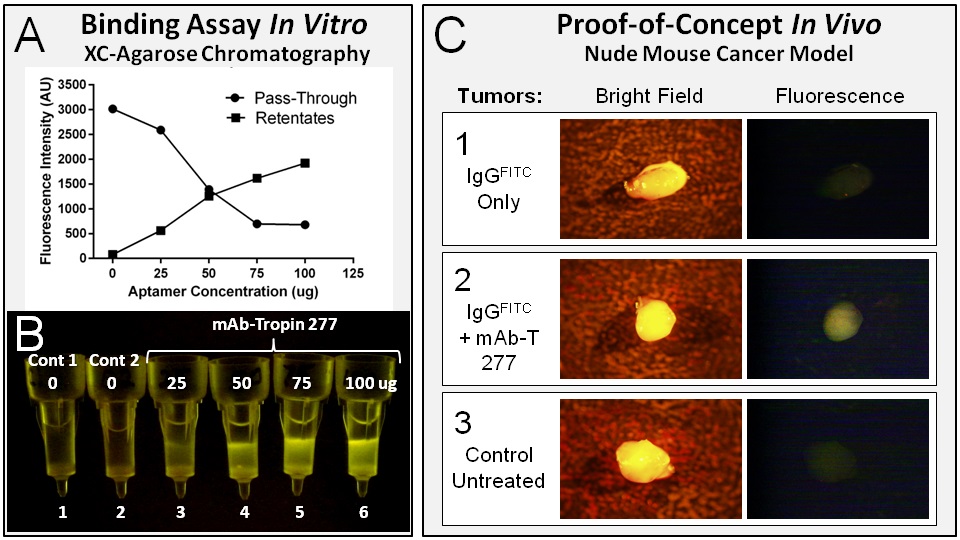
Figure 5: The performance of mAb-Tropin 277 is evaluated in vitro in XC-binding assays and in vivo in a murine xenograft model of metastatic cancer. XC-agarose chromatography using mAb-Tropin 277 and FITC-IgGs, followed by stringent washing and analysis of column eluates and retentates (A, B), demonstrates aptamer-dependent binding of IgGs to XC-proteins (retentates), as well as depletion of the fluorescent IgGs from solution (column pass-through). The mAb-Tropin 277 dependent targeting of fluorescent IgGs (via the systemic circulation) to tumors is demonstrated in a murine metastatic cancer model (C). Taken together with thein vitro binding data which showed that the bifunctional mAb-targeting onco-aptamers efficiently sequester IgGs (A) and concentrate the bound IgGs on collagen matrices (B) this in vivo data indicates that mAb-Tropins concentrate and compartmentalize IgGs/mAbs within the TME (C).
Table 1: Shows the comparative intensity of fluorescence in subcutaneous tumors and normal organs of control and treated mice, designated as positive (+), equivocal (+), or negative (-).
| Mouse Treatment Description | Tumor | Liver | Heart | Muscle | Colon | Bone |
|---|---|---|---|---|---|---|
| Comparison of XC-Probes | ||||||
| Control | - | - | - | ND | ND | ND |
| XC-Probe Bv1 200 ug, 8 min | + | + | - | ND | ND | ND |
| XC-Probe Hs2 200 ug, 8 min | ++ | + | - | ND | ND | ND |
| XC-Probe Hs2 100 ug, 8 min | + | + | + | ND | ND | ND |
| XC-Probe Hs2 200 ug, 8 min | +++ | + | + | + | ND | ND |
| Analysis of mAb-Tropins | ||||||
| Control | - | - | - | - | - | - |
| G0, No Aptamer, 15 min | + | + | - | + | + | - |
| G0, No Aptamer, 15 min | + | - | - | - | + | - |
| G0, No Aptamer, 60 min | + | + | - | - | + | - |
| G0, No Aptamer, 60 min | - | + | - | + | + | + |
| G2, mAb-Tropin 277, 15 min | ++ | - | - | - | - | - |
| G2, mAb-Tropin 277, 15 min | ++ | - | - | - | - | - |
| G2, mAb-Tropin 277, 60 min | ++ | + | - | - | - | - |
| G2, mAb-Tropin 277, 60 min | +++ | - | - | + | - | - |
| Control | - | - | - | - | - | - |
| Control | - | - | - | - | - | - |
| G4, PEG mAb-Tropin, 15 min | + | - | - | - | - | - |
| G4, PEG mAb-Tropin, 15 min | + | - | - | + | - | - |
| G4, PEG mAb-Tropin, 60 min | + | - | - | - | - | - |
| G4, PEG mAb-Tropin, 60 min | + | + | - | + | - | - |
| Note: The selected XC-Probe and Optimal mAb-Tropin (mAb-Tropin 277) are shown in bold print. | ||||||
Comparisons of FITC-conjugated XC-Probes: At 7 minutes, intense fluorescence signals were detected in tumors of mice treated with the XC-Probe Hs2 (200 ug), with slightly less fluorescence intensity observed in tumors of mice treated with Bv1 (200 ug) and Hs2 (100 ug). Fluorescence was not detected in tumor, liver and heart of control untreated mice. Fluorescence was not detected in heart and muscle of mice treated with Hs2 and Bv1, while mild fluorescence was detected in the liver (known site of FITC metabolism/clearance).
Comparisons of FITC-IgGs +/- mAb-Tropins: In pharmacokinetic studies, the time course of appearance and disappearance of fluorescence in tumors and organs of treated mice was determined. At 15 minutes, mild (+) fluorescence signals were detected in tumors of all mice (n = 2) treated with IgG+ phosphate buffered saline (PBS, G0), while more intense (++) signals were noted in tumors of all mice (n = 2) treated with IgG + mAb-Tropin 277 (G2). Minimal fluorescence signal was detected in tumors of mice (n = 2) treated with IgG+ Pegylated (PEG) mAb-Tropin 4 (G4). Persistence of fluorescence signals was noted at 60 minutes, with more intense signals (++, +++) detected in tumors of all mice (n = 2) treated with IgG + mAb-Tropin 277, while mild fluorescence was detected in only 1 of 2 tumors of mice treated with IgG + PBS (G0). Late appearance of mild fluorescence signals (+) at 60 minutes was noted in tumors of all mice (n = 2) treated with IgG + PEG mAb-Tropin 4 (G4). Mild fluorescence signals were detected in the colon of all mice (n = 2) treated with IgG + PBS (G0). In contrast, no fluorescence was noted in the colon of mice (n = 4) treated with IgG + mAb-Tropin 277 and mAb-Tropin 4 (G2 and G4), indicating selective tumor targeting of IgG + mAb-Tropin complexes.
Taken together, these data provide experimental evidence that mAb-Tropins(i) bind tightly to the constant Fc regions of therapeutic mAbs after simple mixing prior to infusion; (ii) retain the therapeutic bioactivity in the process of this high-affinity non-covalent mAb binding; (iii) seek out exposed XC-proteins of the ECM within the TME; and (iv) accumulate in high localized concentrations within the tumors while sequestering the complexes away from normal non-target cells and tissues, thereby compartmentalizing both mAb biodistribution and bioactivity in the TME.
Summary and Conclusion
The concept of targeting XC-proteins, which are a characteristic histopathology of all invasive cancers, has evolvedinto an enabling platform biotechnology for tumor-targeted drug delivery, both historically, in terms of enabling tumor-targeted gene delivery [19-25], and presently, for targeting therapeutic mAbs more selectively and efficiently to tumor compartments while sparing non-target organs and tissues of untoward side effects. Based on previous designs of targeted growth factors and genetic medicines, the vWF-derived tumor-targeting sequences were further refined by comparative binding assays performed in vitro using XC-agarose chromatography, with the optimal sequences confirmed in vivo in animal models of metastatic cancer. Optimal IgG-binding sequences, structural orientations, linker sequences, and N-terminal and C-terminal modifications to extend circulating half-life were also determined experimentally. A chromatographic simulation of histological tumor-targeting in vitro can be seen in Figure 2: The XC-targeting mAb-Tropin 277, bound non-covalently to FITC-labeled IgGs, is shown to be selective for XC-agarose (a proxy for tumor tissues) over control agarose (normal tissues). Moreover, the pharmacokinetic advantages of active mAb-Tropin-dependent tumor targeting over passive IgG/mAb accumulations are demonstrated in an animal model of metastatic pancreatic cancer.
The most advanced onco-aptamer design (i.e. mAb-Tropin 277) is a bifunctional fusion polypeptide combining [i] a proven tumor-targeting function with [ii] a high-affinity IgG-binding property, to generate stable drug-aptamer complexes with improved biodistribution. When added to the final monoclonal antibody formulation by simple mixing just prior to infusion, the mAb-Tropin binds tightly to the therapeutic IgG/mAb and guides the antibody-aptamer complex through the general circulation to primary and metastatic lesions by seeking out abnormal XC-proteins of the TME, thereby increasing the effective local mAb concentrations within the tumors. These essential proofs-of-principle could lead to the design of new clinical trials using mAb-Tropin 277 to target FDA-approved chemotherapeutic agents, anti-angiogenic or immune checkpoint inhibitors.
Competing Interests
NSC- no competing interest. FLH and EMG are co-inventors of patents covering bifunctional fusion polypeptides, and FLH, SPC and EMG are founders and non-paid officers of Counterpoint Biomedica LLC.
Authors’ Contributions
FLH conceived of the study, designed the experiments, performed the in vitro binding assays, developed the chromatographic tumor simulation experiments, analyzed the data, and revised the manuscript. EMG helped design the experiments, performed the in vivo animal studies, and helped analyze the data, drafted the manuscript, and revised the manuscript. SPC helped design the experiments and analyze the data, reviewed the manuscript draft, and revised the manuscript. NSC helped design the experiments and analyze the results, helped draft the manuscript and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgment
The authors are grateful to the staff of Anticancer Inc., San Diego CA, for their assistance in the conduct of the in vivo experiments, and to Heather C. Gordon for graphic illustrations and editing assistance in the writing of this manuscript.
Abbreviations: mAbs-Monoclonal antibodies; irAEs-Immune-related adverse events; TME-Tumor microenvironment; ECM-Extracellular matrix; XC proteins-Exposed collagenous proteins; vWF-Von Willebrand factor; FDA-Food and Drug Administration; IgG-Immunoglobulin G; FITC-Fluorescein isothiocyanate; VEGF-Vascular endothelial growth factor; PBS-Phosphate buffered saline; PEG-Pegylated; CTLA-4-Cytotoxic T-lymphocyte associated protein; PD-1-Programmed cell death protein-1; PD-L1-Programmed cell death protein ligand-1; Fmoc-9-fluorenylmethyloxycarbonyl; HPLC-High-performance liquid chromatography; DMSO-Dimethyl sulfoxide; HUVEC-Human umbilical vein endothelial cells; bFGF-Basic fibroblast growth factor; FBS-Fetal bovine serum.
References
- 1. Oldham, R.K., Dillman, R.O. Monoclonal antibodies in cancer therapy: 25 years of progress. (2008) J Clin Oncol 26(11):1774-1777.
- 2. Scott, A.M., Wolchok, J.D., Old, L.J. Antibody therapy of cancer. (2012) Nature Rev Cancer. 12:278-87.
- 3. Baldo, B.A. Adverse events to monoclonal antibodies used for cancer therapy Focus on hypersensitivity responses. (2013)Oncoimmunology 2(10): e26333.
- 4. Momtaz, P., Postow, M.A. Immunologic checkpoints in cancer therapy: focus on the programmed death-1(PD-1) receptor pathway. (2014) Pharmgenomics Pers Med 7: 357-365.
- 5. Kri, C., Postow, M.A. Checkpoint blocking antibodies in cancer immunotherapy. (2014) FEBS Letters 588(2):368-376.
- 6. Postow, M.A., Callahan, M.K., Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. (2015) J ClinOncol. 33:1-9.
- 7. Voskens, C.J., Goldinger, S.M., Loquai, C., et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. (2013) PLos One 8(1):e53745.
- 8. Kong, Y.M., Flynn, J.C. Opportunistic autoimmune disorders potentiated by immune-heckpoint inhibitors anti-CTLA-4 and anti-PD-1. (2014)Front Immunol 5: 206.
- 9. Villadolid, J., Amin, A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. (2015) Transl Lung Cancer Res 4(5): 560-575.
- 10. Kim, J.W., Eder, J.P. Prospects for Targeting PD-1 and PD-L1 in Various Tumor Types. (2014) Oncology3:15-28.
- 11. Sznol, M., Chen, L.Antagonist Antibodies to PD-1 and B7-H1 (PD-L1) in theTreatment of Advanced Human Cancer.(2013) Clin Cancer Res 19:1021-1034.
- 12. Hanahan, D., Weinberg, R.A. Hallmarks of cancer: the next generation. (2011) Cell 144:646-674.
- 13. Pietrasa, K., Ostmanb, A. Hallmarks of cancer: Interactions with the tumor stroma. (2010) Exp Cell Res 316(8):1324-1331.
- 14. Pickup, M.W., Mouw, J.K., Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. (2014) EMBO Rep 15:1243-1253.
- 15. Ahmed, F., Steele, J.C., Herbert, J.M., et al. Tumor stroma as a target in cancer. (2008) Curr Cancer Drug Targets; 8(6):447-453.
- 16. Fan,F., Schimming, A., Jaeger, D., et al. Targeting the tumor microenvironment: focus on angiogenesis. (2012) J Oncol 2012: 281261.
- 17. Sounni, N.E., Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin Chem 59(1): 85-93.
- 18. Bryckaert, M., Rosa, J.P., Denis, C.V., et al. Of von Willebrand factor and platelets. (2015) Cell Mol Life Sci 72(2): 307-326.
- 19. Hall, F.L., Liu, L., Zhu, N.L., et al. Molecular engineering of matrix-targeted retroviral vectors incorporating a surveillance function inherent in von Willebrand factor. (2000) Hum Gene Ther 11(7): 983-993.
- 20. Gordon, E.M., Hall, F.L. Noteworthy clinical case studies in cancer gene therapy: tumor-targeted Rexin-G advances as an efficacious anti-cancer agent. (2010) Int J Oncol 36(6): 1341-1353.
- 21. Chawla, S.P., Chua, V.S., Fernandez, L., et al. Phase I/II and phase II studies of targeted gene delivery in vivo: intravenous Rexin-G for chemotherapy-resistant sarcoma and osteosarcoma. (2009) Mol Ther 17(9):1651-1657.
- 22. Chawla, S.P., Chua, V.S., Fernandez, L., et al. Advanced phases I/II studies of targeted gene delivery in vivo: intravenous Rexin-G for gemcitabine-resistant metastatic pancreatic cancer. (2010) Mol Ther 18(2): 435-441.
- 23. Hall, F.L., Levy, J.P., Reed, R.A., et al. Pathotropic targeting advances clinical oncology: tumor-targeted localization of therapeutic gene delivery. (2010) Oncol Rep24:829-833.
- 24. Waehler, R., Russell, S.J., Curiel, D.T. et al. Engineering targeted viral vectors forgene therapy. (2007)Nat Rev Genet 8(8): 573-587.
- 25. Gordon, E.M., Hall, F.L. Rexin-G, a targeted genetic medicine for cancer. (2010) Expert Opin Biol Ther 10(5): 819-832.










