
The Fate of Human Lung Epithelial Cells - Death or Survival Induced by Sodium Arsenite
Yongping Mu1,2, Dao-Joanna Nguyen1,3, He Wang1,4*
Affiliation
- 1Department of Environmental Health Science, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA 70112, USA
- 2Clinical Laboratory Tests Center, The Affiliated People’s Hospital of Inner Mongolia Medical University, Inner Mongolia Autonomous Region, 010010, China
- 3Eleanor McMain Secondary School, New Orleans, LA 70128, USA
- 4School of Health Sciences, University of Newcastle, Callaghan, NSW 2308, Australia
Corresponding Author
He Wang, School of Health Sciences, University of Newcastle, Callaghan, NSW2308, Australia, Tel: (02) 49217735; E-mail: he.wang@newcastle.edu.au
Citation
Wang, H., et al. The Fate of Human Lung Epithelial Cells – Death or Survival Induced by Sodium Arsenite. (2017) J Med Chem Toxicol 2(2): 95- 99.
Copy rights
© 2017 Wang, H. Sams. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Abstract
Arsenic as a toxic metalloid has been found to have potential carcinogenic effects of humans. However, pulmonary toxicity has not been fully elucidated. The purpose of this experiment is to test and observe the degree of toxicity of various concentrations of sodium arsenite (NaAsO2) on human lung epithelial cells (BEAS-2B cell line). In this experiment, BEAS-2B cells were plated and grown according to experimental requirements and treated with different concentrations of NaAsO2 for various exposure durations to study the biological responses of the human lung cells. MTT assay and ROS (reactive oxygen species) production were used to observe and visualize the effects of cytotoxicity on cell viability of BEAS-2B cells. Necrotic cell death was measured by lactic dehydrogenase (LDH) level. Western Blotting was used to detect LC3B-I and LC3B-II (autophagy related protein), cleaved PARP (apoptosis related protein), and surviving (apoptosis inhibitor related protein) to understand the biological responses of BEAS-2B cells after NaAsO2 exposure. This study supports that acute, in vitro exposure to NaAsO2 on BEAS-2B cells induced concentration-dependent decrease in cell viability and increase in ROS generation in BEAS-2B. For necrosis, it occurred at high concentrations of arsenite indicating the resistance of BEAS-2B to arsenite toxicity. It also supports that NaAsO2 exposure induces both autophagy and apoptosis, but autophagy occurred at much lower concentrations than apoptosis did in the arsenite-exposed BEAS-2B.
Introduction
Arsenic, a toxic metalloid, has been found to have potential carcinogenic effects of humans[1]. It is widespread in the environment today and can be found in necessary human resources, including: drinking water, food, air, and fuel sources[2,3]. Because avoiding exposure to arsenic has become so difficult, understanding the toxicity of exposure amounts and durations to arsenic has become a major focus of research. The purpose of this experiment is to observe the biological response of BEAS-2B cells when exposed to various concentrations of sodium arsenite (NaAsO2). The production of ROS is associated with altering various cellular signaling pathways that regulate and promote cell survival[4] as well as regulate cell death[5,6]. ROS generation is increased under stressful conditions[7]. ROS-induced DNA damage and PARP are required for induction of stress induced autophagy. Autophagy, a cellular self-degrading process that balances sources of energy in stressful conditions, is important for maintaining homeostasis in cell[8]. During autophagy, a cytosolic form of light chain 3 (LC3; LC3-I) is cleaved and then conjugated to phosphatidylethanolamine to form the LC3-phosphatidylethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes[9]. When cells are exposed to tolerable stress conditions, autophagy occurs as a method of repair and cell proliferation. However, when cells are exposed to severe or extreme stress conditions, autophagy may not be sufficient enough for survival and will trigger apoptosis, which is called programmed cell death[10]. Acute cell death that is called necrotic cell death. Poly (ADP-ribose) (PAR) polymerase (PARP) enzymes catalyze the conversion of NAD+ to polymers of PAR[11] which role in the DNA damage response has long been recognized. Recent works indicate that PAR itself acts to directly induce cell death through stimulation of apoptosis-inducing factor release[12].
Materials and Methods
Reagents and materials
Trivalent Inorganic Arsenic (Sodium (meta) arsenite [NaAsO2], MW 129.91, abbreviated as iASIII (purity > 98%), phenylmethylsulfonyl fluoride (PMSF), lactic dehydrogenase (LDH) and protease inhibitor cocktail were supplied by Sigma-Aldrich (St. Louis, MO). Trypsin/1mM EDTA (0.25%), DMEM medium, 1 × PBS, NP40 cell lysis buffer, and penicillin/streptomycin were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Lawrenceville, GA). BCA assay kit was obtained from Pierce Biotechnology (Rockford, IL). BEAS-2B cells were purchased from the ATCC (Catalog No. CRL - 9609™). The following primary polyclonal antibodies used in this experiment were purchased from Cell Signaling Technology (Boston, MA): LC3B, PARP, surviving and β-actin (or GAPDH). IRDye® 800CW-conjugated secondary antibody was purchased from BioRad (Life Science Research 2000 Alfred Nobel Drive Hercules, CA 94547).
Preparation of sodium arsenite solution
Trivalent Inorganic Arsenic (sodium arsenite) was dissolved in double-distilled water to a final concentration of 10−2 M. This stock solution was freshly prepared for every test. Serial dilutions were prepared starting from the stock solution to obtain different arsenite concentrations (0 μmol/l - 20 μmol/l) in cell media.
Cell culture
The BEAS-2B cells were cultured in a complete medium, consisting of DMEM medium, supplemented with 10% fetal bovine serum (FBS), 100 μg /ml penicillin and 100 g/ml streptomycin and maintained at 37°C and 5% CO2 in a humidified incubator. BEAS-2B cells were treated with each NaAsO2 concentration for 12h, 24h and 48h.
Analysis of cell viability by MTT assay
MTT assay was used to evaluate the viability of BEAS-2B cells cultured in media containing different concentrations of sodium arsenite (0.00, 0.3125, 0.625, 1.25, 2.50, 5.00, 10.0, 20.0 μmol/l) according to the instruction of the manufacturer’s instruction. Briefly, 1.00 x 105 of BEAS-2B cells were seeded into 96-well plates overnight to adhere, and the media containing various concentrations of sodium arsenite was added to the wells. After incubation for 12h, 24h and 48h, MTT 5 mg/ml, equal of 1/10 of media, was added to each well and incubated for 3 h. After incubation, the medium was removed, and equal media of DMSO was added to the remaining precipitates on the plates in the well. The absorbance of the resulting solution was determined at 570 nm (690 nm wavelength was used as reference) using a spectrophotometer (BIO-RAD).
Reactive oxygen species (ROS) assay
BEAS2B cells treated with different concentrations of NaAsO2 for 48h, washed, incubated with Dichloro dihydro-fluorescein diacetate (DCFH-DA) for 45 min. The fluorescent intensity was measured using a multi detection microplate reader (FLUO star Optima Microplate Reader, BMG LABTECH, Cary, NC) with excitation and emission wavelengths of 485 nm and 520 nm, respectively. Three independent experiments were conducted.
Western blot assay
To determine the relative protein expression, total proteins were extracted and analyzed by the immunoblot assay. Cells were harvested following indicated time exposure to various concentrations of arsenite. The lysates were sonicated and centrifuged for 5 minutes at 10,000 x g and supernatant fractions were stored at – 80°C or used immediately. The total protein extracts were separated by 6% - 12% SDS-PAGE gel and immunoblot analysis was performed according to the manufacturer’s instruction (Cell signaling, USA), after electro-transferring, membrane was blocked in TBS / 0.1% Tween 20 with 5% (w/v) nonfat dry milk for 1 hour at room temperature, then incubated with primary polyclonal antibodies overnight at 4°C. After washing with TBS/0.1% Tween 20, target protein band, specifically bound to the primary antibody, was detected by LI-COR (LI-COR Odyssey, USA) system with IR Dye® 800CW-conjugated secondary antibody. Western blot bands were quantified by densitometry relative to the GAPDH loading control using image software.
Statistical analysis
All experiments were repeated at least three times. The data were expressed as means ± SD. (standard deviation). Intergroup differences were tested by analysis of variance (One Way ANOVA), followed by Tukey’s test. The software used was Excel and SPSS, version 13.0 (SPSS Inc., Chicago, IL, USA). Values of p < 0.05 were considered statistically significant (indicated in tables and figures as *).
Results
When BEAS-2B cells were treated with increasing concentration of NaAsO2 for 12h, 24h, 48 h exposing BEAS-2B cells to a low concentration of 0.3125 μM of NaAsO2 at all three tested time durations (12h, 24h, 48h) actually promotes cell proliferation. However, as the concentration increases (0.625 μM – 20μM) and as the time of exposure increases, cell viability begins to dramatically decrease (Figure 1).
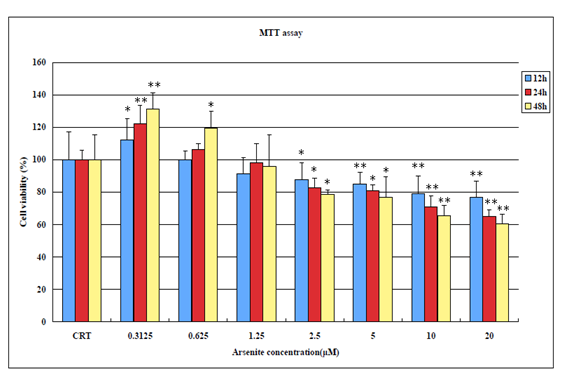
Figure 1: MTT Assay results showing cell viability of BEAS-2B cells after exposure to NaAsO2 for 12h, 24h and 48h (*p < 0.05; **p < 0.01 versus control).
Unexposed BEAS-2B grow adhered to the bottom of the plate at high confluence whereas NaAsO2 treated cells begin to die and float at low concentrations, and at a relatively high concentration of 5μM, cells begin to float and develop a significant change in morphology. An observation consistent with decreased cell viability in the MTT assays (Figure 2). In Figure 3, ROS production increases in a NaAsO2 concentration dependent manner at 24h and 48h exposure. For necrosis, it occurred at high concentrations of arsenite (10 μM -20 μM) indicating the resistance of BEAS-2B to arsenite toxicity.

Figure 2: BEAS_2B cell morphology changes relative to different concentrations (0 μM - 20 μM) of NaAsO2 after 24 hrs of treatment.
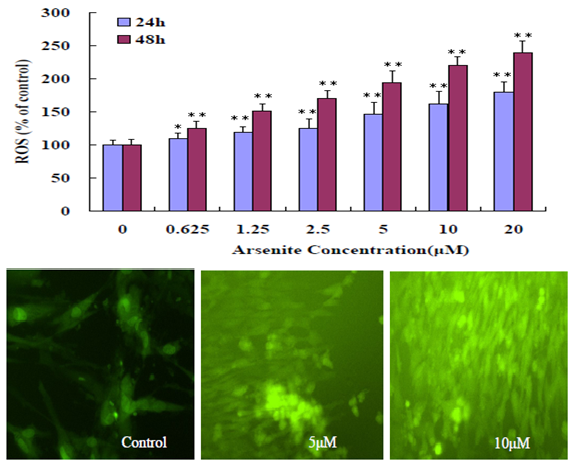
Figure 3: Induction of oxidative stress in BEAS-2B cells. The upper figure shows BEAS-2B cells treated with different concentration (0 - 20 μM) NaAsO2 for 24h, 48h. The results represented the mean ± SD of three independent experiments. (*P < 0.05, **P < 0.01, compared with untreated cells 24h, 48h). The lower figure shows fluorescent intensity increased with different concentration (0 - 20 μM) NaAsO2 for 48h.
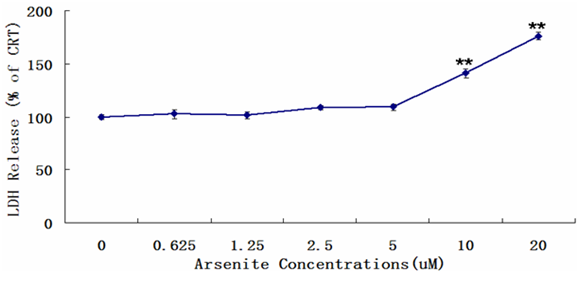
Figure 4: Release of LDH in BEAS-2B exposed to sodium arsenite for 24h. The data show the levels of lactate dehydrogenase increase in a concentration manner in BEAS-2B. LDH release was measured by microplate reader at 450 nm wavelength. Results were presented as mean ± SD of three independent experiments (*P < 0.05, **P < 0.01, compared with untreated cells, control group).
An indication of autophagy occurrence in cells is the change of the expression ratio of LC3-II: LC3-1. LC3-II is a specific marker for the quantity of autophagosomes and autolysosomes[13]. Autophagy increases the ratio of LC3-II to LC3-I due to increased conversion of LC-3-I to LC3-II. As shown in Figure 5, the ratio of LC3-II to LC3-I steadily went up as NaAsO2 concentration is increased from zero to 1.25 μM, providing additional evidence of autophagy occurring after exposure to NaAsO2, but begins to decrease at a relatively high concentration of 2.5μM (Figure 6).
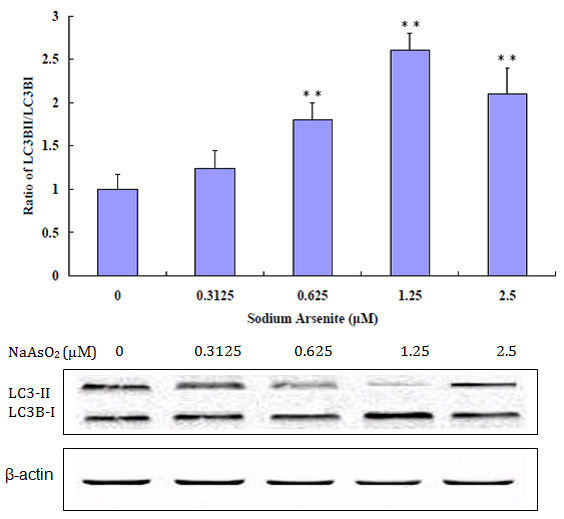
Figure 5: Western Blot analysis for LC3B-I and LC3B-II. Increased ratio of LC3B-II and LC3B-I at 0 μM to 1.25 μM NaAsO2 concentrations. Decreased ratio of LC3B-II and Lc3B-I at a relatively high concentration of 2.5 μM. (**p < 0.01 versus control).
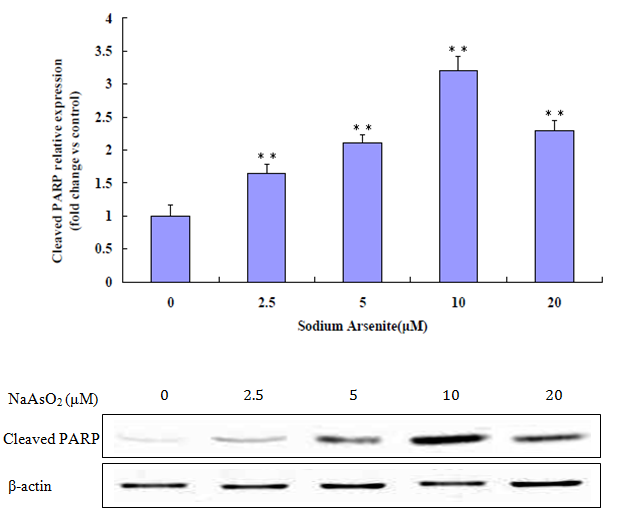
Figure 6: Western Blot analysis for cleaved PARP. Increased cleaved PARP at 0 μM to 10 μM. Decreased at a relatively high concentration of 20μM. (**p < 0.01 versus control).
The lung epithelial cells treated with NaAsO2 showed increased expression of active PARP, a hallmark protein of apoptosis. Results shown in (Figure 7) confirmed that the active 89kDa cleaved PARP was increasingly expressed in cells exposed to progressively higher NaAsO2 concentrations. Figure 6 shows that at 0 μM to 1.25 μM concentrations of NaAsO2 protein expression of surviving increases, but at concentrations of 2.5 μM to 20 μM, expression of surviving decreases.
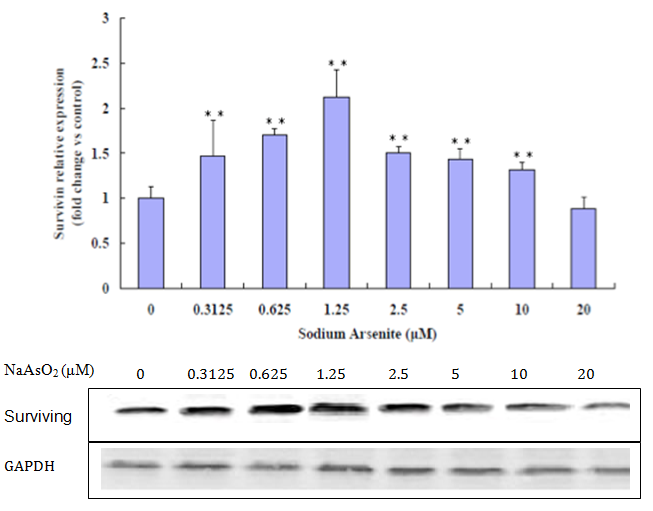
Figure 7: Western Blot analysis for surviving. Increased surviving at 0 μM to 1.25 μM. Decreased at a relatively high concentration of 2.5 μM (**p < 0.01 versus control).
Discussion and Conclusion
Cellular accumulation of ROS play a vital role in the stimulation of autophagy under conditions of cellular stress responses, such as nutrient deficiency[14], Oxidative stress[7], ROS are unstable molecules. There are two major forms of ROS (H2O2 and O2−), both of which mediate the induction of autophagy[14].
This study supports that sodium arsenite induces oxidative stress in BEAS-2B cells. Based on the ROS production assay completed, increases in NaAsO2 exposure levels were found to increase ROS production. This increase in ROS generation by NaAsO2 contributes to cell toxicity on the BEAS-2B cell line. This experiment also supports that exposing BEAS-2B cells to NaAsO2 in vitro induces autophagy and apoptosis. A low concentration exposure to NaAsO2 is found to result in autophagy, which is supported by protein expression of LC3B-I and LC3B-II (autophagy related proteins) at 0 μM to 1.25 μM. Based on the decrease of LC3B-I and LC3B-II and on the increase of cleaved PARP (apoptosis related protein) at a relatively high concentration of 2.5 μM NaAsO2, it supports that apoptosis is induced at higher concentrations of NaAsO2. The increase in surviving, an inhibitor of apoptosis, at low concentrations (0 μM to 1.25 μM) and decrease at a relatively high concentration (2.5 μM to 20 μM) also support that apoptosis occurs at high concentrations.
Autophagy is an evolutionarily conserved cellular catabolic process in which proteins and organelles are eliminated through delivery to lysosomes and serves as a cell survival mechanism under oxidative stress conditions[15]. Autophagy (type 2 cell death) is an adaptation and survival pathway but if these conditions are maintained for a long time or extreme, autophagy may not be sufficient enough for survival, excessive autophagy will trigger apoptosis , can lead to cell death, often called programmed cell death (PCD, type 1 cell death)[16]. PCD is morphologically defined as a type of cell death that occurs in the presence of nuclear fragmentation, chromatin condensation, plasma membrane blebbing, caspase activation or engulfment by phagocytes in vivo[15]. Level of LDH in BEAS-2B exposed to sodium arsenite for 24h was detected. The data show the levels of lactate dehydrogenase increase in high concentration (over 5 μM) in BEAS-2B.
surviving (BIRC5), a member of the inhibitor of apoptosis protein family that inhibits caspases and blocks cell death is highly expressed in cancer and is associated with a poorer clinical outcome[17]. Functioning simultaneously during cell division and apoptosis inhibition, surviving plays a pivotal role in determining cell survival[18]. surviving increases at low concentrations of arsenite, expression of surviving decreases at relative high concentrations of arsenite in the experiment.
All together, these data indicate that continuous exposure to sodium arsenite has biological effects, such as autophagy and apoptosis. Autophagy may be cytoprotective, at least under conditions of oxidative stress and point to a potential cross talk between type 1 and type 2 cell deaths on lung epithelial cells. This suggests that long term exposure to NaAsO2 by air or drinking water can have significant damaging impacts on human lungs and overall health.
References
1. Cui, X., Kobayashi, Y., Akashi, M., et al. Metabolism and the paradoxical effects of arsenic: carcinogenesis and anticancer. (2008) Current medicinal chemistry 15(22): 2293-2304.
2. Nordstrom, D.K. Public health. Worldwide occurrences of arsenic in ground water. (2002) Science (New York, NY) 296(5576): 2143-2145.
3. Garelick, H., Jones, H., Dybowska, A., et al. Arsenic pollution sources (2008) Reviews of environmental contamination and toxicology 197: 17-60.
4. Salmeen, A., Andersen, J.N., Myers, M.P., et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. (2003) Nature 423(6941): 769-773.
5. Boya, P., Gonzalez-Polo, R.A., Casares, N., et al. Inhibition of macroautophagy triggers apoptosis. (2005) Molecular and cellular biology 25(3): 1025-1040.
6. Yang, C., Kaushal, V., Shah, S.V., et al. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. (2008) American journal of physiology Renal physiology 294(4): F777-787.
7. Wang, F., Zhou, X., Liu, W., et al. Arsenite-induced ROS/RNS generation causes zinc loss and inhibits the activity of poly (ADP-ribose) polymerase-1. (2013) Free radical biology & medicine 61: 249-256.
8. Yorimitsu, T., & Klionsky, D.J. Autophagy: molecular machinery for self-eating. (2005) Cell death and differentiation 12 Suppl 2: 1542-1552.
9. Kabeya, Y., Mizushima, N., Ueno, T., et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. (2000) The EMBO journal 19(21): 5720-5728.
10. Baehrecke, E. H. Autophagy: dual roles in life and death? (2005) Nature reviews Molecular cell biology 6(6): 505-510.
11. Schreiber, V., Dantzer, F., Ame, J.C., et al. Poly (ADP-ribose): novel functions for an old molecule. (2006) Nature reviews Molecular cell biology 7(7): 517-528.
12. Wang, Y., Dawson, V.L., Dawson, T.M. Poly (ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. (2009) Experimental neurology 218(2):193-202.
13. Korolchuk, V.I., Menzies, F.M., Rubinsztein, D.C. A novel link between autophagy and the ubiquitin-proteasome system. (2009) Autophagy 5(6): 862-863.
14. Hariharan, N., Zhai, P., Sadoshima, J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. (2011) Antioxidants & redox signaling 14(11): 2179-2190.
15. Levine, B., Kroemer, G. Autophagy in the pathogenesis of disease. (2008) Cell 132(1): 27-42.
16. Kroemer, G., Levine, B. Autophagic cell death: the story of a misnomer. (2008) Nature reviews Molecular cell biology 9(12): 1004-1010.
17. Altieri, D. C. surviving, cancer networks and pathway-directed drug discovery. (2008) Nature reviews Cancer 8(1): 61-70.
18. Li, F., Ambrosini, G., Chu, E. Y., et al. Control of apoptosis and mitotic spindle checkpoint by survivingg. (1998) Nature 396(6711): 580-584.










