
Traumatic Brain Injury Causes a Tacrolimus-Sensitive Increase in Non-Convulsive Seizures in a Rat Model of Post-Traumatic Epilepsy
John N. Campbell, Anandh Gandhi, Baljinderjit Singh, Severn B. Churn
Affiliation
- 1Anatomy and Neurobiology, Virginia Common Wealth University, Richmond, VA
- 2Neurology, Virginia Commonwealth University, Richmond, VA
- 3Physiology and Biophysics, Virginia Common Wealth University, Richmond, VA
- 4Pharmacology and Toxicology, Virginia Common Wealth University, Richmond, VA
- #These authors contributed equally to this work
Corresponding Author
Severn B. Churn, Virginia Common Wealth University, Richmond, Virginia 23298, Post Box-980599,Tel: (804) 828 0290; E-mail: schurn@vcu.edu
Citation
Severn B. C., et al. Traumatic Brain Injury Causes a Tacrolimus-Sensitive Increase in Non-Convulsive
Copy rights
©2014. This is an Open access article distributed under the terms of Creative Commons Attribution 4
Keywords
Post-traumatic epilepsy; Epileptogenesis; FK-506; EEG; Tonic-clonic seizure; Absence seizure
Abstract
Epilepsy is a significant but potentially preventable complication of traumatic brain injury (TBI). Previous research in animal models of acquired epilepsy has implicated the calcium-sensitive phosphatase, calcineurin. In addition, our lab recently found that calcineurin activity in the rat hippocampus increases acutely after lateral TBI. Here we use a calcineurin inhibitor test whether an acute increase in calcineurin activity is necessary for the development of late post-traumatic seizures. Adult rats were administered the calcineurin inhibitor Tacrolimus (5mg/kg; i.p.) 1 hour after lateral fluid percussion TBI and then monitored by video-electrocorticography (video-ECoG) for spontaneous seizure activity 5 weeks or 33 weeks later. At 5 weeks post-TBI, we observed epileptiform activity on the video-ECoG of brain injured rats but no seizures. By 33 weeks post-TBI though, nearly all injured rats exhibited spontaneous seizures, including convulsive seizures which were infrequent but lasted minutes (18% of injured rats), and non-convulsive seizures which were frequent but lasted tens of seconds (94% of injured rats). We also identified non-convulsive seizures in a smaller subset of control and sham TBI rats (56%), reminiscent of idiopathic seizures described in other rats strains. Non-convulsive seizures in the brain injured rats, however, were four-times more frequent and two-times longer lasting than in their uninjured littermates. Interestingly, rats administered Tacrolimus acutely after TBI showed significantly fewer non-convulsive seizures than untreated rats, but a similar degree of cortical atrophy. The data thus indicate that administration of Tacrolimus acutely after TBI suppressed non-convulsive seizures months later.
Introduction
Traumatic brain injury (TBI) occurs every 19 seconds in the United States[1] and results in chronic, recurrent seizures in least 1 in 50 of those who survive[1,2]. This form of acquired epilepsy, known as post-traumatic epilepsy, is often refractory to medications or surgery[3,4] and so can be a lifelong disability for the victim and a financial burden which extends to their community. An ideal therapy would prevent or limit the development of post-traumatic epilepsy, but no such therapy has yet been found[5]. A better understanding of how the injured brain becomes epileptic, a process termed epileptogenesis, should reveal novel opportunities for preventing post-traumatic epilepsy.
Epileptogenesis is thought to begin within minutes to hours after brain injury[6,7], though the underlying mechanisms are unknown. One hypothesis is that a pathological increase in intracellular calcium triggers molecular changes which culminate in neuronal hyper excitability[8,9]. Calcium-sensitive enzymes which regulate neuronal excitability may thus play a critical role in epileptogenesis. Indeed, animal models of acquired epilepsy have implicated a calcium-sensitive phosphatase, calcineurin[9-12]. Recent research by our lab suggests that calcineurin may also be involved in the epileptogenesis after TBI. In a study of lateral fluid percussion TBI in rats, a well-characterized model of post-traumatic epilepsy[13,14], we identified an acute post-injury increase in calcineurin activity[15]. This increase could facilitate epileptogenesis through the dysregulation of hyperpolarizing cyclic nucleotide-gated (HCN) channels[10] or voltage-gated potassium channels (KV2.1)[16]. An increase in calcineurin activity may also alter excitatory synaptic circuits by destabilizing dendritic spines[17-22], potentially creating opportunities for maladaptive plasticity[23]. The increase in calcineurin activity we observed after TBI could therefore be an early but critical step in epileptogenesis.
The present study characterized the importance of calcineurin activity in a rat model of post-traumatic epilepsy. We administered the calcineurin inhibitor, Tacrolimus (FK506), to adult rats 1 hour after lateral fluid percussion TBI and then monitored spontaneous seizure activity 5 weeks or 33 weeks later. The results indicate a significant disease-modifying effect of Tacrolimus in this model of post-traumatic epilepsy.
Materials and Methods
Ethics statement
All animal care and use complied strictly with the Guide for the Care and Use of Laboratory Animals as described by the National Institutes of Health, and was approved in advance by the Virginia Commonwealth University Institutional Animal Care and Use Committee (approval number, AM10270). All efforts were made to minimize animal suffering and distress.
Animal use and housing conditions
The present study involved a total of 49 male, Sprague-Dawley rats acquired as adults (90 day old) and randomly assigned to treatment groups in two experiments. Experiment 1 assessed spontaneous seizure activity at 33 weeks post-TBI and involved the following treatment groups: controls (naïve, age-matched rats; n=7), sham TBI (n=3), TBI Only (n=10), and TBI+TAC (n=14). Experiment 2 examined spontaneous seizure activity at 5 weeks post-TBI in different rats assigned to the same treatment groups: control (naïve, age-matched rats; n=4), TBI Only (n=5), and TBI+TAC (n=6). Throughout the experiment, all animals received ad libitum access to food and water and were kept in temperature- and humidity-controlled housing on 12-hour light/dark cycles.
Lateral fluid percussion injury
The procedure for lateral fluid percussion injury has been described previously[15,17] and was identical for Experiments 1 and 2. All rats except controls were each anesthetized with 3% isoflurane in a carrier gas mixture of 30% N2O and 70% O2 and placed into a stereotactic frame. A 5mm circular craniotomy was made over the left hemisphere using a manual trephine centered between Lambda and Bregma (approximately -4.4mm Bregma), and midway between the sagittal suture and the lateral ridge. A modified syringe hub was placed over the craniotomy, affixed to the skull with a cyanoacrylate adhesive, and then secured with dental acrylic. The hub was filled with sterile saline and the scalp was sutured closed over the hub. On the following day, the same rats were anesthetized with isoflurane (4min of 4% isoflurane in a carrier mixture of 30% N2O and 70% O2) and subjected to fluid percussion of the intact dura over their left parietal cortex. Sham TBI rats received the same dose of isoflurane but no fluid percussion. The fluid percussion device used in Experiment 1 is identical to that described by Dixon and colleagues[17]. A newer model of this device was used in Experiment 2. Fluid percussion pressure was measured at the time of fluid impact, by an external pressure transducer adjacent to the hub. The Luer-Loc fitting, screw, and dental cement were removed from the skull and the scalp was sutured closed immediately after fluid percussion injury (or, in sham TBI rats, immediately after discontinuation of isoflurane anesthesia). If apnea was observed following injury, rats were mechanically ventilated with room air, beginning within 10 seconds of apnea onset and continuing until spontaneous respiration resumed. Immediately following injury, rats were placed in a supine position and the time at which they righted themselves was recorded. Body temperature was maintained for at least 1 hour after injury by placement on a heating pad set to "low." Rats were then returned to their home cages and monitored daily. Some rats (TBI+TAC) received a single injection of Tacrolimus (5mg/kg; intraperitoneal, i.p.; Astellas Pharma Inc., Tokyo, Japan) 1 hour after TBI.
Video-electrocorticography
Each rat was anesthetized with 3% isoflurane in a carrier gas mixture of 30% N2O and 70% O2. The scalp was then incised and reflected, and surgical steel screws with Teflon-coated leads (1.6mm long shafts, 10mm long leads; Plastics One, Norfolk, VA, U.S.A.) were implanted over the frontal cortex (3.5mm anterior to Bregma, +/- 2.5mm lateral to midline), the right parietal cortex (2.0mm posterior to Bregma, +2.5mm lateral to midline) and the left parietal/occipital cortex (8.0mm posterior to Bregma, -2.5mm lateral to midline). To protect underlying brain tissue, electrode screws were inserted into the skull to a depth of no more than 0.8mm (i.e., half of the electrode screw length). Next, the tips of each electrode lead were inserted into a plastic electrode platform (Plastics One), the entire headset was then secured in place with dental acrylic, and the scalp was sutured closed around the base of the headset. Rats were then allowed to recover for at least 3 days prior to EEG recording, after which they were lightly sedated with isoflurane (i.e., to the point of slowing reflexes) and connected to a video-ECoG system (BMSI 5000, Nicolet, Middleton, WI, U.S.A.). A bipolar montage was used, with individual traces representing the voltage difference between (from top trace to bottom) the two frontal leads, the two ipsilateral leads, the two contralateral leads, and the two posterior leads. In Experiment 1, video-ECoG was recorded for at least 12 hours per rat. In Experiment 2, video-ECoG was recorded daily, or every other day, until at least 40 hours were recorded in total per rat. To ensure the quality of video data for behavioral analysis, recordings were scheduled so that at least two-thirds of video would be recorded during light hours.
Analysis of spontaneous seizures (Experiments 1 and 2)
Video-ECoG data in Experiment 1 were analyzed offline by three identically-trained reviewers, one of whom was blinded to treatment groups. In Experiment 2, all video-ECoG data were analyzed offline by a single reviewer who was blinded to treatment groups. All ECoG traces were visually reviewed for electrographic seizures, defined as paroxysmal, rhythmic trains of spikes, waves, or spike-waves with amplitude at least double that of baseline (i.e., the 3-second epoch preceding the event) and a duration of 5 seconds or longer. The duration of ECoG discharge was interpreted as seizure duration. To characterize ECoG power distribution, spectral analysis with Insight II software was performed on a representative sample of seizures (200Hz sampling of ≥3 second epochs; Persyst Corporation, Prescott, AZ). To estimate the average frequency of seizures (seizures per hour) for each rat, the number of seizures observed on video-ECoG was divided by the number of video-ECoG hours reviewed. Seizure behavior was scored by comparison to a modified Racine scale[24]: sudden freezing was scored as a "0," facial twitching as a "1," head bobbing as a "2," forelimb clonus as a "3," rearing as a "4," and falling or jumping as a "5." Seizures with a Racine score of 0 to 2 were considered "non-convulsive," whereas those with a Racine score of 3 to 5 were identified as "convulsive." Seizures were characterized as "non-convulsive" (Racine stage 0 to 2) or "convulsive" (Racine stage 3 to 5) according to the corresponding behavior.
Histology and analysis of neocortical volume (Experiments 1 and 2)
Rats were deeply anesthetized with a euthanasia cocktail (390mg/kg of pentobarbital, 50mg/kg of phenytoin; i.p.) and then transcardially perfused with room-temperature 0.9% saline followed by phosphate-buffered 4% paraformaldehyde (Pease's fixative). Brains were extracted whole and then immersed in Pease's fixative overnight at 4°C. Brains were examined grossly for evidence of electrode injury, typically appearing as a small (<1mm) brown lesion on the surface of the neocortex underlying an electrode. Any rats showing such injury were excluded from all analyses. Brains were blocked to remove tissue rostral to Bregma and caudal to Lambda. Tissue blocks were then sectioned in the coronal plane by vibratome (Leica, Wetzler, Germany). Sections were cut slowly and at a thickness of 75μm to minimize fraying and folding of peri-lesional tissue. Beginning at the hippocampal commissure (-0.8mm Bregma), every eighth section was retained until 9 sections were collected per brain (1-in-8 series, 600μm intervals; last section at -5.6mm Bregma). This range permitted analysis of tissue located 3.6mm rostral and 1.2mm caudal to the site of fluid percussion impact. Slide-mounted sections were stained with thionine, dehydrated in ethanol, cleared in xylene, and cover slipped with Per mount (Thermo Scientific). Sections were then imaged through a macro lens (0.5x) on a light microscope (Nikon Eclipse E800M). Images were opened in ImageJ[25], which was then used to trace the borders of the ipsilateral and contralateral neocortex and measure the enclosed area. The area of the ipsilateral neocortex was then divided by that of the contralateral neocortex, yielding a ratio ("RatioCx") for each brain section[26]. An analogous procedure was used to calculate a ratio of the hippocampal hemispheres (RatioHp). Regional boundaries and distances from Bregma were defined according to a stereotaxic atlas[27].
Statistical analysis
The normality of data distribution was assessed by Shapiro Wilk test (α = 0.05). Normal data were compared by paired Student's t test, unpaired Student's t test, or one-way analysis of variance (ANOVA) with Tukey's post hoc test (for paired, single, or multiple comparisons, respectively), including the following: FPI pressure, subject age, and post-TBI time. Non-normal data were compared by Wilcoxon matched-pairs signed rank test, Mann Whitney test, or Kruskall-Wallis test with a post hoc Mann Whitney test (for paired, single, or multiple comparisons, respectively), including the following: righting reflex latency; seizure frequency; seizure duration; total seizure time; RatioCx; and RatioHp. Acute mortality rate, prevalence of seizures, and prevalence of thalamic calcifications were compared across groups using a Chi-square test. Spearman's test was used to assess the correlation between seizure frequency and each histological measure (RatioCx, RatioHp, and thalamic calcifications). When testing for a correlation between seizure frequency and thalamic calcifications, each rat was scored as a "1" or "0" depending on the presence or absence of calcifications, respectively. All statistical analyses were performed with an alpha level of 0.05, using GraphPad Prism (version 5.0; GraphPad Software, La Jolla, CA, U.S.A.). Data are reported as mean ± standard deviation (SD) in the text but are illustrated as mean ± standard error of the mean (SEM) in the figures. N values represent the number of rats, unless stated otherwise.
Results
The present study used a total of 49 male, Sprague-Dawley rats acquired as adults (90 days old) and randomly assigned to treatment groups in two experiments. Rats were subjected to traumatic brain injury (TBI) by lateral fluid percussion and then monitored for spontaneous seizure activity at either 33 weeks post-TBI (Experiment 1) or 5 weeks post-TBI (Experiment 2).
Mortality and severity of lateral fluid percussion injury
To ensure that TBI severity was comparable between Experiments 1 and 2, we examined two correlates of TBI severity, the latency to recovery of righting reflex after TBI ("righting time") and the acute mortality rate (i.e., within 7 days of TBI). Overall, the average righting time was 9.1 ± 3.8 minutes and the average mortality rate was 14%. Neither righting time nor acute mortality rate differed significantly between experiments (righting time, p=0.4599; acute mortality rate, Χ2 [5]=3.461, p=0.6293), thus indicating that TBI severity was equivalent between the two experiments.
Experiment 1: Adult, male Sprague-Dawley rats (n = 34; 14 ± 1 weeks old) were administered lateral fluid percussion TBI or a sham injury (n = 3), or were kept as age-matched controls (n = 7). At 1 hour post-TBI, rats received either a single dose of the calcineurin inhibitor, Tacrolimus (5mg/kg; i.p.; "TBI+TAC"; n = 14) or no injection ("TBI Only"; n = 10). Age at the time of TBI (or sham) did not differ significantly among treatment groups (F[2,16]= 0.4307, p = 0.6574; Table 1). Three rats died within 7 days of TBI (acute mortality rate, 13%), including one rat humanely sacrificed due to severe motor impairment. Acute mortality rate varied across treatment groups but not to a statistically significant extent (mortality rates by treatment: TBI Only, 20%; TBI+TAC, 7%; Χ2[2]=0.8816, p = 0.6435). The rats which died acutely after TBI were excluded from all analyses, as were the additional two rats that died after 7 days post-TBI but before data collection. Three of the surviving rats (TBI+TAC group) exhibited apnea immediately following TBI and were mechanically ventilated with room air until spontaneous respirations resumed (mean duration of ventilation, 1.8 ± 1.1 minutes). Fluid percussion pressure (surviving rats) and righting time (all rats) were measured as correlates of injury severity. Among the rats surviving TBI and used for video-ECoG analysis, fluid percussion pressure averaged 2.7 ± 0.1 atmospheres and righting time averaged 9.5 ± 4.2 minutes (Table 1). Neither fluid percussion pressure nor righting time differed significantly between TBI Only and TBI+TAC rats (fluid percussion pressure, t[15] = 1.495, p = 0.1558; righting time, p = 0.5622), which demonstrates that TBI severity was equivalent between these groups.
Experiment 2: Adult, male Sprague-Dawley rats (n = 15; 14 ± 1 weeks old) were administered lateral fluid percussion TBI, followed 1 hour later by either Tacrolimus (n = 6; TBI+TAC) or no injection (n = 5; TBI Only). Uninjured littermates were kept as age-matched controls (n = 4; Control). Two rats in the TBI+TAC group died within 7 days of TBI (acute mortality rate, 33%) and so were excluded from analysis. All other rats survived until completion of the experiment. Overall, the acute mortality rate associated with TBI in Experiment 2 was 18%. Fluid percussion pressure and acute mortality were again used to approximate injury severity. The mean fluid percussion pressure among surviving rats was 2.36 ± 0.04 atmospheres, and the mean righting time was 8.3 ± 2.8 minutes (Table 1). Neither injury measure differed significantly between TBI Only and TBI+TAC rats (fluid percussion pressure, t[7]=1.562, p=0.1622; righting time, p = 0.9048), indicating that the severity of TBI was equivalent between these groups.
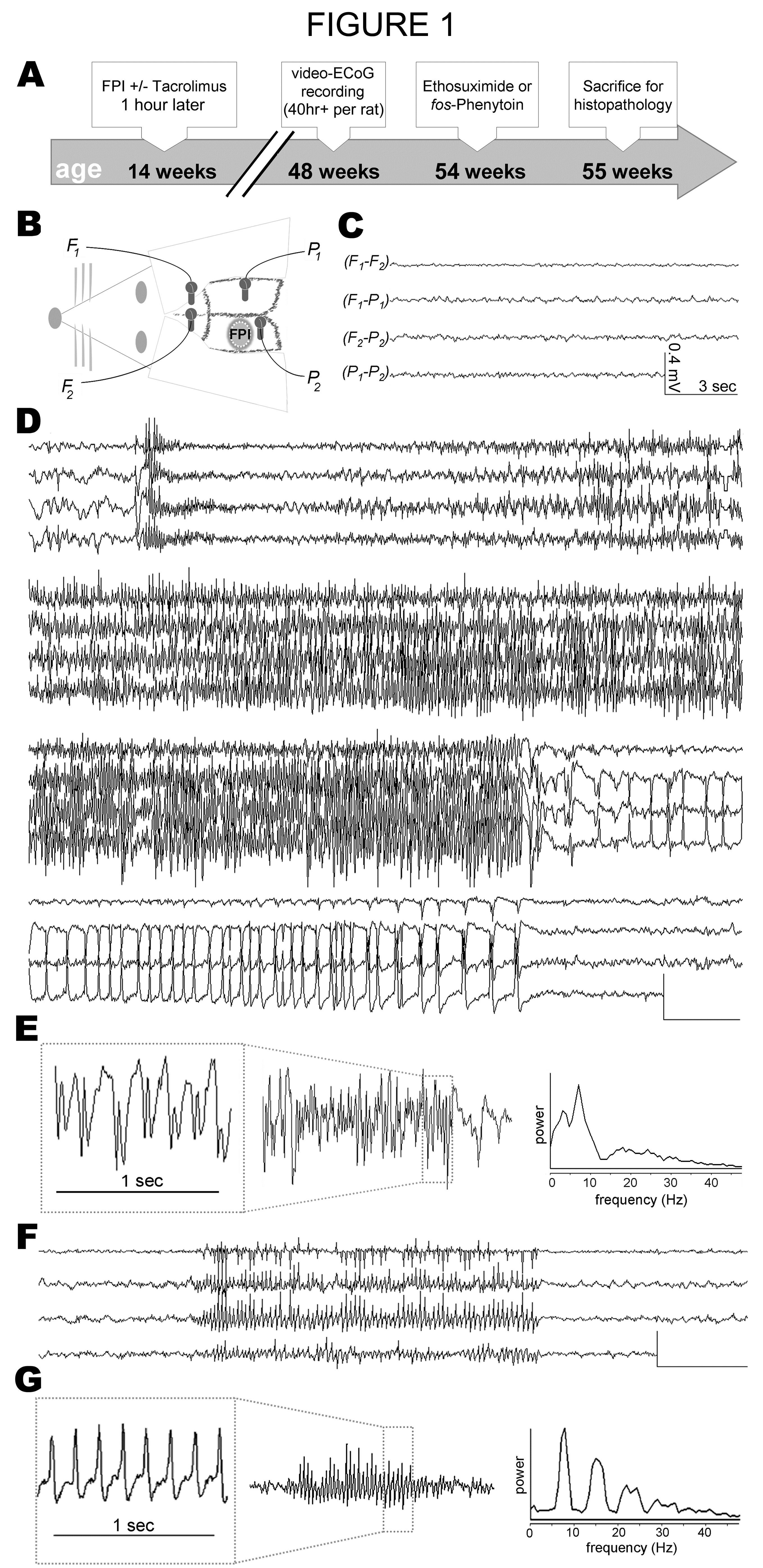
Figure 1:
Video-electrocorticography revealed two distinct types of late post-traumatic seizures (Experiment 1): Video-Electrocorticography Revealed Two Distinct Types of Late Post-Traumatic Seizures: Adult, male Sprague-Dawley rats were subjected to lateral fluid percussion TBI or sham TBI and then monitored for spontaneous seizure activity 33 ± 5 weeks later, alongside age-matched uninjured controls. A) Timeline of Experiment 1. B) Placement of fluid percussion impact (FPI) and epidural electrodes for electrocorticography (ECoG). C) Arrhythmic, low-voltage ECoG activity typical of awake behavior in all rats, shown as bipolar montage. D) Representative ECoG of convulsive (tonic-clonic) seizures observed months after TBI. E) On left, enlarged ECoG trace showing irregular spike-and-wave waveform of convulsive seizures. On right, spectral analysis of a 3 second epoch showing typical distribution of power during a convulsive seizure. F) Representative example of ECoG of non-convulsive seizure. G) On left, enlarged ECoG trace revealing spike-wave waveform of non-convulsive seizures. On right, spectral analysis of a 3 second epoch showing typical distribution of power during a non-convulsive seizure. All scale bars, 0.4mV by 3sec.
Video-Electrocorticography Revealed Two Distinct Types of Late Post-Traumatic Seizures: Adult, male Sprague-Dawley rats were subjected to lateral fluid percussion TBI or sham TBI and then monitored for spontaneous seizure activity 33 ± 5 weeks later, alongside age-matched uninjured controls. A) Timeline of Experiment 1. B) Placement of fluid percussion impact (FPI) and epidural electrodes for electrocorticography (ECoG). C) Arrhythmic, low-voltage ECoG activity typical of awake behavior in all rats, shown as bipolar montage. D) Representative ECoG of convulsive (tonic-clonic) seizures observed months after TBI. E) On left, enlarged ECoG trace showing irregular spike-and-wave waveform of convulsive seizures. On right, spectral analysis of a 3 second epoch showing typical distribution of power during a convulsive seizure. F) Representative example of ECoG of non-convulsive seizure. G) On left, enlarged ECoG trace revealing spike-wave waveform of non-convulsive seizures. On right, spectral analysis of a 3 second epoch showing typical distribution of power during a non-convulsive seizure. All scale bars, 0.4mV by 3sec. We used simultaneous video-ECoG to monitor spontaneous seizure activity at 33 ± 5 weeks after TBI or sham, or at the equivalent age in control rats (Figure 1A & 1B). One rat (sham TBI) was excluded from analysis due to a surgical injury of the neocortex which occurred during implantation of the ECoG electrodes. Age at time of video-ECoG did not differ significantly among treatment groups (F[3,22] = 2.539; p = 0.0827; Table 1). On average, video-ECoG was recorded over the course of 8 ± 7 days for a total of 51 ± 15 hours per rat (Table 1).
Table 1: TBI Severity, Age, and Video-ECoG Duration: Values represent mean ± standard deviation. Note that the difference in FPI pressure between Experiments 1 and 2 is most likely due to the use of different FPI devices.
| Exp. # | Treatment Group | N | FPI pressure (atm) | Righting time (min) | Age at TBI/sham (weeks) | Age at video-ECoG (weeks) | Age at sacrifice (weeks) | Video-ECoG record (hours) |
|---|---|---|---|---|---|---|---|---|
| 1 | Control | 7 | n/a | n/a | n/a | 43.2 ± 8.1 | 43.8 ± 8.3 | 42.4 ± 7.9 |
| sham | 2 | n/a | n/a | 15.0 ± 0.6 | 45.0 ± 0.0 | 62.1 ± 0.0 | 43.7 ± 4.3 | |
| TBI only | 6 | 2.74 ± 0.11 | 9.2 ± 5.0 | 14.0 ± 0.7 | 50.8 ± 4.2 | 56.2 ± 2.3 | 58.6 ± 28.3 | |
| TBI+TAC | 11 | 2.67 ± 0.07 | 9.7 ± 4.0 | 14.1 ± 1.6 | 45.6 ± 3.1 | 56.6 ± 9.3 | 52.6 ± 5.6 | |
| 2 | Control | 4 | n/a | n/a | n/a | 19.8 ± 0.1 | 19.9 ± 0.1 | 10.8 ± 0.7 |
| TBI only | 5 | 2.34 ± 0.04 | 8.5 ± 3.7 | 13.0 ± 0.0 | 17.9 ± 0.1 | 17.9 ± 0.1 | 11.7 ± 0.9 | |
| TBI+TAC | 6 | 2.38 ± 0.03 | 8.2 ± 1.4 | 14.0 ± 0.6 | 19.2 ± 0.5 | 19.4 ± 0.4 | 12.3 ± 0.3 |
In one case the ECoG headset detached from a rat after only two days of recording, limiting that rat's ECoG record to 12.4 hours. We confirmed electrode depth by examining the neocortex post-mortem. Two rats (TBI Only and TBI+TAC) were each found to have a small (<1mm) neocortical lesion at an electrode site and so were excluded from all analyses. The remaining 1,315 hours of ECoG data were visually reviewed for electrographic seizures, defined as paroxysmal, rhythmic trains of spikes, waves, or spike-waves, lasting 5 seconds or longer and with an amplitude at least double that of baseline ECoG activity. All rats exhibited arrhythmic, low-amplitude ECoG activity during awake behavior (Figure 1C). In sharp contrast to this baseline activity were observed two distinct types of electrical seizures. One seizure type appeared on the ECoG as a burst of high-voltage spikes, followed by low-voltage fast activity which evolved into high-voltage spikes and waves (Figure 1D & 1E). Spectral analysis of these seizures showed dominant power in the delta (<4Hz) and theta (4 to 8Hz) ranges (Figure 1E). Analysis of the corresponding video revealed a tonic contraction followed by forelimb clonus, rearing, and falling (i.e., progression from Racine stage 3 to 5; see representative example in Supplementary Video 1) - we therefore classified these events as "convulsive seizures." Previous studies of lateral fluid percussion TBI have reported similar seizures[14,26]
supplementary video 1 - convulsive seizure observed months after tbi
The present study identified convulsive seizures in two rats (TBI+TAC) during video-ECoG recording months after TBI. These seizures occurred at a rate of 0.05 ± 0.04 per hour and lasted an average of 95 ± 14 seconds (n=5 seizures; range 78 - 110 seconds). A third rat (TBI Only) exhibited a convulsive seizure prior to video-ECoG, at 13 weeks post-TBI, but no additional seizures during video-ECoG. Overall, only 5 convulsive seizures were recorded, rarity which precluded further analysis of these seizures.
The second type of seizure we observed was distinct in terms of electrographic appearance, spectral profile, and behavioral correlate. This type of seizure appeared on the ECoG as a rhythmic train of spike-waves, beginning abruptly or building-up rapidly across all four ECoG leads, and lasting for tens of seconds (Figure 1F). These discharges had a spike frequency of 6 to 8Hz, peak spectral power in the theta (4 to 8Hz) and beta (12 to 18Hz) ranges (Figure 1G), and could be interrupted by alerting stimuli such as sudden changes in lighting or noise (data not shown). The corresponding video showed subtle but stereotyped changes in behavior, ranging from behavioral arrest (or immobility) to head bobbing (Racine stage 0 to 2; see representative example in Supplementary Video 2) - we termed these events "non-convulsive seizures." Similar seizures have been described in studies of adult rats following lateral[13] or parasagittal fluid percussion TBI[13,28,29] or neonatal hypoxic injury[30]
Supplementary video 2 - Non-Convulsive seizure observed months after TBI
We identified spontaneous, non-convulsive seizures at 33 weeks post-TBI in 94% rats (16 of 17), prevalence similar to that observed months after parasagittal fluid percussion[28,29]. One rat (TBI Only) did not exhibit any non-convulsive seizures on video-ECoG, only a single convulsive seizure which occurred prior to video-ECoG recording (see above). The two other rats with convulsive seizures also exhibited non-convulsive seizures, though convulsive and non-convulsive seizures were never observed on the same day in the same rat. A total of 3,279 non-convulsive seizures were identified in the video-ECoG record of TBI rats.
Previous ECoG studies have reported idiopathic seizure activity in uninjured Sprague-Dawley rats, typically beginning around 7-8 months of age (e.g.,[28,29]). Accordingly, the present study identified non-convulsive seizures in 3 of 7 control rats and 2 of 2 sham TBI rats (average age, 10 ± 2 months). The seizures in control and sham TBI rats were similar electrographically, characterized by a generalized onset, spike-wave morphology, and peak spectral power in the theta and beta ranges (Figure 2A). The corresponding video showed behavioral freezing (or immobility) lasting at least as long as the electrographic discharge.
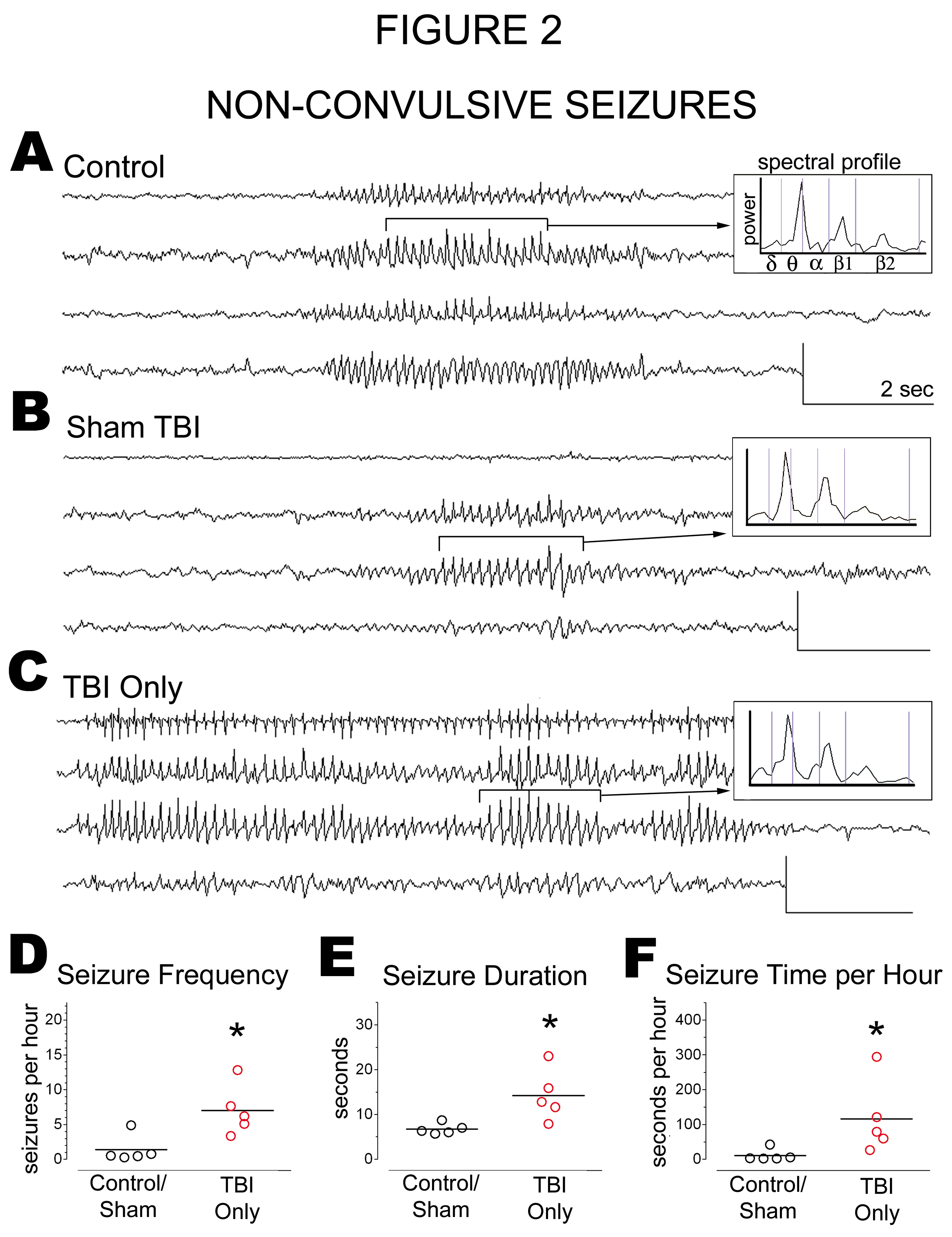
Figure 2:
Late Post-Traumatic Seizures Could Be Distinguished from Idiopathic Seizures by Frequency and Duration: Video - ECoG was performed on male Sprague-Dawley rats 33 weeks after lateral fluid percussion TBI ("TBI Only") or sham ("Sham TBI"), or at the equivalent age in uninjured littermates ("Control"). A - C) Representative ECoG and spectral profiles of non-convulsive seizures in an uninjured control rat, a Sham TBI rat, and a TBI Only rat. D, E) Non-convulsive seizures were significantly more frequent and longer lasting in TBI Only rats than in controls/shams. F) TBI Only rats spent on average significantly more time in a non-convulsive seizure than did controls/shams. Circles and bars on graphs denote individual means and group means, respectively. Means compared by Kruskal-Wallis test with post hoc Mann Whitney tests. Scale bars, 0.4mV by 2 seconds. *, p < 0.05
TBI increased the frequency and duration of non-convulsive seizures
The non-convulsive seizures of age-matched control and sham TBI rats resembled those of TBI rats (Figure 2A-2C). We therefore compared groups in terms of seizure prevalence, calculated as the percentage of rats in which at least 1 seizure was observed during video-ECoG monitoring. Non-convulsive seizures appeared to be more prevalent in TBI Only rats (83%) than in control and sham TBI rats (56%, combined), though the difference was not statistically significant (χ2[1] = 0.8365, p = 0.3604).
We also compared groups in terms of seizure frequency (Figure 2D). Data from control and sham TBI rats were pooled for this analysis, since these groups did not differ significantly in seizure frequency (control, 2.1 ± 2.5 seizures per hour; Sham TBI, 0.4 ± 0.1 seizures per hour; t[3] = 0.9146, p = 0.4279). Controls and sham TBI rats together exhibited 1.4±1.9 non-convulsive seizures per hour (n = 243 seizures). The frequency of non-convulsive seizures was 5-fold higher in TBI Only rats than in controls/shams (TBI Only rats, 7.0 ± 3.6 non-convulsive seizures per hour; n = 1,713 seizures; TBI Only vs. controls/shams, t[8] = 3.075, p = 0.0152).
We then compared seizure duration across groups (Figure 2E). Data from control and sham TBI rats were again pooled, since seizure duration did not differ significantly between these groups (control, 7 ± 1 seconds; sham TBI, 6 ± 0 seconds; t[3] = 1.656, p = 0.1963). The mean duration of non-convulsive seizures in controls/shams was 6.8 ± 1.2 seconds. The durations of non-convulsive seizures in TBI Only rats was twice as long as in controls/shams (13.6 ± 11.6 seconds; t[8] = 2.887, p = 0.0203; Figure 2E). In addition, the longest non-convulsive seizure recorded in controls/shams was 15 seconds, but in TBI Only rats was 181 seconds.
An increase in seizure frequency and duration could mean that more time per hour is spent in a seizure state. To test this possibility, we multiplied the averages of seizure frequency and seizure duration for each rat. This analysis indicated that TBI Only rats averaged ten-fold more time per hour in a seizure state than did controls/shams (p = 0.0159; Figure 2F). Thus, non-convulsive seizures in TBI Only rats were significantly more frequent and longer-lasting than in controls/shams.
Tacrolimus administered at one hour post-tbi reduced the frequency of non-convulsive seizures months later
Our lab recently demonstrated that calcineurin activity in the rat cortex increases within hours of lateral fluid percussion TBI[15], suggesting that calcineurin may be involved in post-traumatic epileptogenesis. We therefore administered a calcineurin inhibitor, Tacrolimus (5mg/kg; i.p.; TBI+TAC; n = 12) to rats 1 hour after lateral fluid percussion TBI and then monitored the rats by video-ECoG alongside their untreated counterparts (TBI Only) at 33 ± 5 weeks post-TBI. As mentioned above, 1 TBI+TAC rat was excluded from analysis due to a small (<1mm) neocortical lesion at an electrode site. Data from the remaining TBI+TAC rats (n = 11) are summarized below.
Like the TBI Only rats, TBI+TAC rats exhibited spontaneous non-convulsive seizures months after lateral TBI (Figure 3A). The prevalence of these seizures did not differ significantly between TBI+TAC rats and TBI Only rats (100% and 83%, respectively; χ2[1] = 1.948, p = 0.1628). We then compared TBI groups in terms of seizure frequency and duration. This analysis indicated that non-convulsive seizures were significantly less frequent in the TBI+TAC group than in the TBI Only group (p = 0.0414; Figure 3B). However, seizure duration did not differ significantly between the TBI groups (p = 0.2127; Figure 3C). Multiplying seizure frequency by seizure duration for each rat revealed that TBI+TAC rats averaged significantly less seizure time per hour than did TBI Only rats (p = 0.0414; Figure 3D). The data overall indicate that administering Tacrolimus at 1 hour post-TBI reduced the frequency of spontaneous non-convulsive seizures months later.
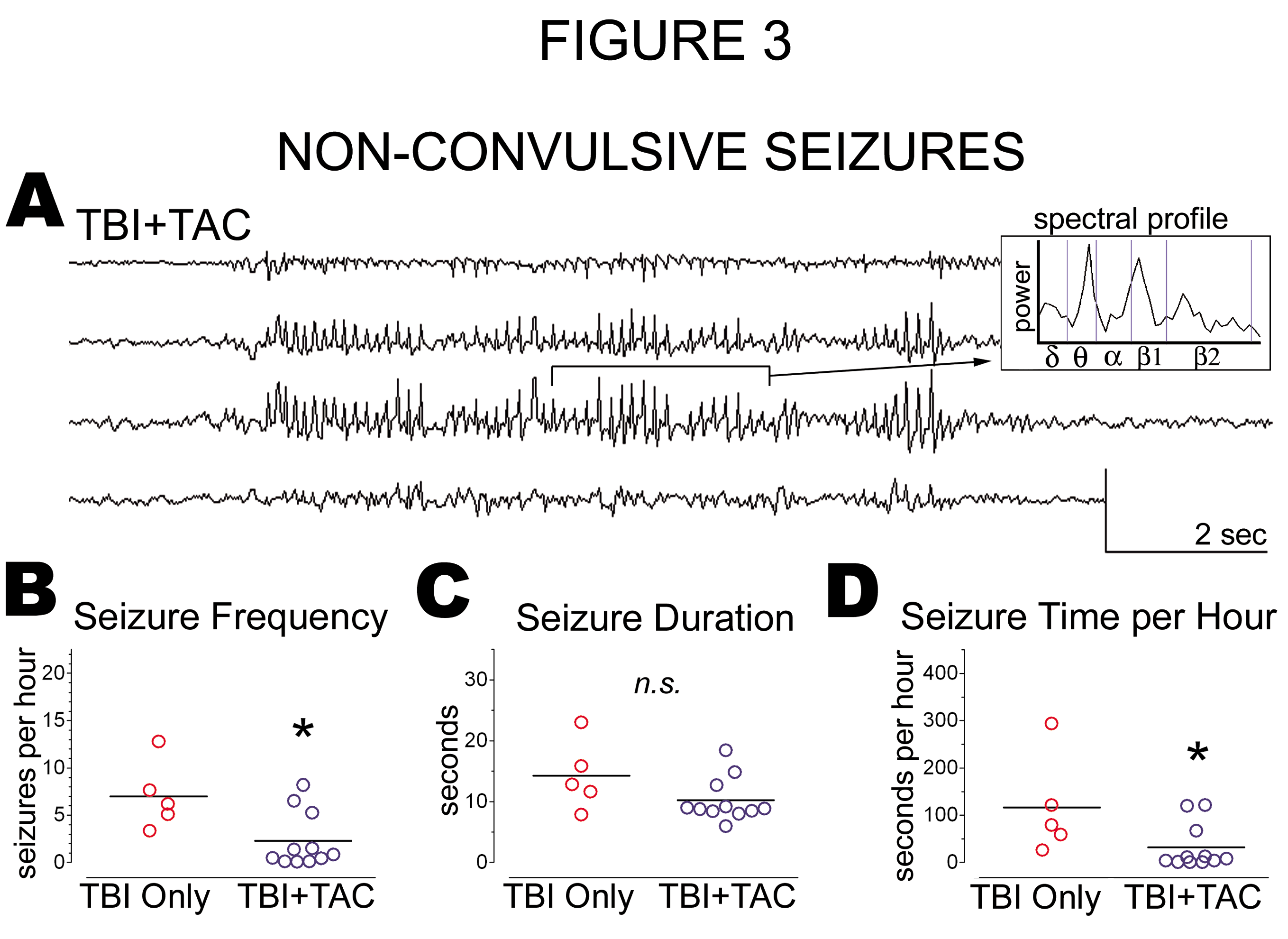
Figure 3:
Tacrolimus Administered Acutely after TBI Decreased the Frequency of Late Non-Convulsive Seizures: Rats were administered a calcineurin inhibitor, Tacrolimus (5mg/kg; i.p.; "TBI+TAC") or no injection ("TBI Only") 1 hour after lateral fluid percussion TBI, and then monitored by video-ECoG approximately 33 weeks later. A) A typical non-convulsive seizure in a TBI+TAC rat and its spectral profile. B) TBI+TAC rats exhibited significantly fewer non-convulsive seizures per hour than did TBI Only rats. C) The average duration of non - convulsive seizures did not differ significantly between TBI+TAC and TBI Only rats. D) TBI+TAC rats averaged significantly less time per hour in a non-convulsive seizure than did TBI Only rats. Circles and bars on graphs denote individual means and group means, respectively. Means compared by Kruskal-Wallis test with post hoc Mann Whitney tests. All scale bars, 0.4mV by 2 seconds. * p < 0.05
Differences in seizure frequency between groups could be explained by differences in the light/dark dependency of seizures. For example, since most video-ECoG was recorded during light hours, seizure frequency could be underestimated if more seizures occurred during the dark hours. Plotting seizure onset times on a Raster did not suggest any light/dark dependence (Figure 4A), and seizure frequency did not differ significantly between light hours and dark hours of ECoG recording (for each treatment group, p > 0.05 for light vs. dark seizure frequency; paired Student's t test; Figure 4B).
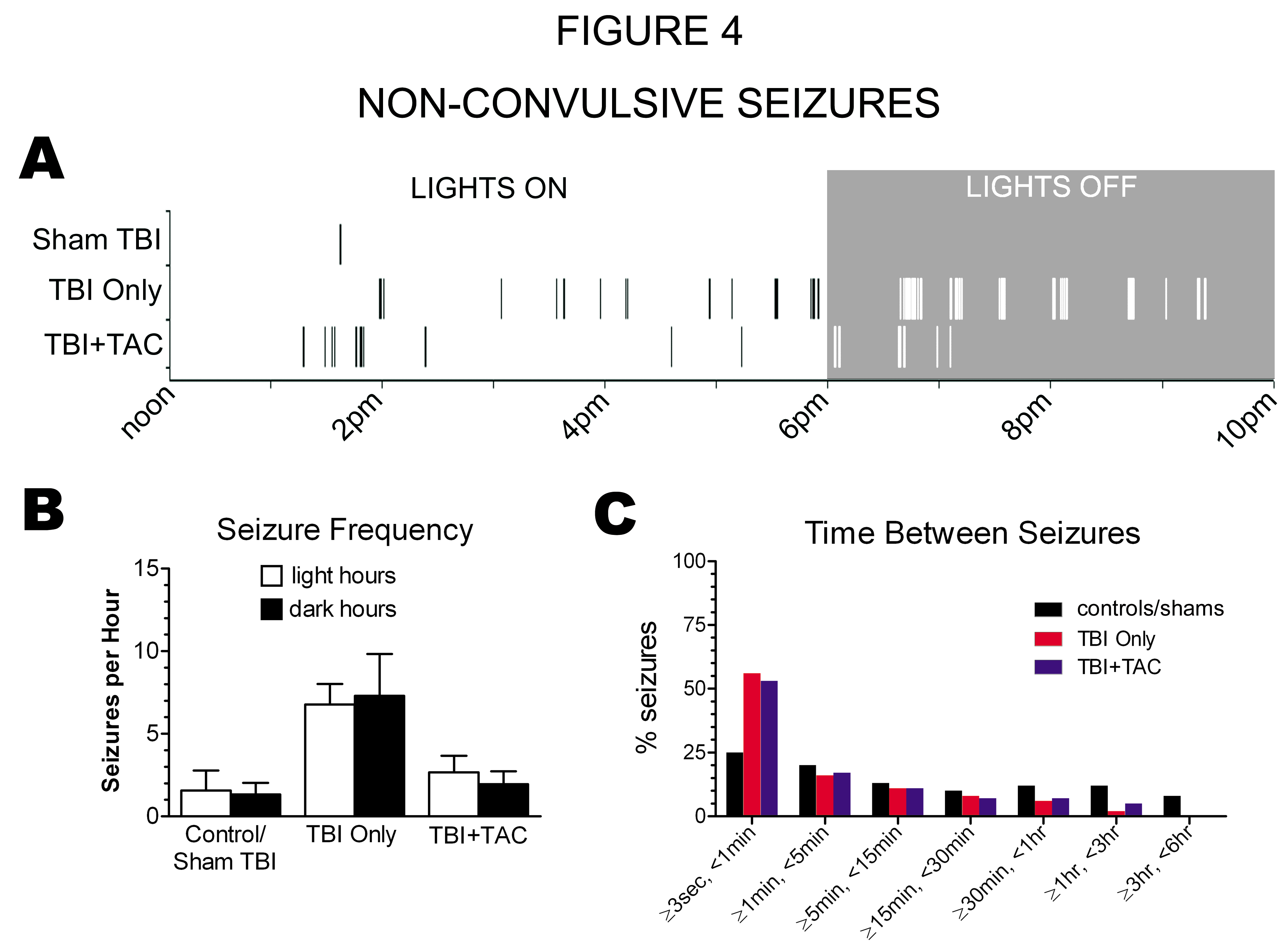
Figure 4:
Late Non-Convulsive Seizures Occurred in Clusters: Adult, male Sprague-Dawley rats were administered lateral fluid percussion TBI followed by Tacrolimus ("TBI+TAC") or no injection ("TBI Only"), and then were monitored for spontaneous seizure activity months later, along with age-matched uninjured ("Controls") and Sham TBI rats. A) Raster plot with hashes (|) marking the onset times of spontaneous non-convulsive seizures during typical 10 hour video-ECoG recordings of Sham TBI, TBI Only, and TBI+TAC rats. B) Seizure frequency did not differ significantly between light and dark phases (paired Student's t tests). C) Binned analysis revealed that a majority of non-convulsive seizures occurred within seconds of one another in TBI Only rats and TBI+TAC rats, though not in controls/shams.
Review of the Raster plots also revealed a clustering pattern to the non-convulsive seizures, especially in the brain injured rats (Figure 4A). To confirm the apparent clustering, we averaged the time between seizures for each rat and then compared group means (Figure 4C). A binned analysis indicated that about 50% of non-convulsive seizures occurred within seconds of one another in the TBI Only and TBI+TAC rats. In the controls/shams however, only about 25% of non-convulsive seizures occurred within seconds of one another. The apparent increase in seizure clustering in TBI rats could be due to an increase in seizure frequency. However, seizure frequency did not differ significantly between the TBI+TAC and controls/shams (p>0.05), though seizures tended to cluster more in the TBI+TAC rats (Figure 4C). The data thus suggest that TBI enhances clustering of non-convulsive seizures, independent of the change in seizure frequency.
Nearly one-third of the ECoG data in Experiment 1 were reviewed blind to treatment group (28% overall; by treatment group, 0% control/sham, 34% TBI Only, and 43% TBI+TAC). The remaining data were reviewed without blinding and so may have been subject to experimenter bias. To control for this potential confound, we tested whether blinding affected seizure identification in the ECoG reviewers. Three ECoG recordings (~18 ECoG hours total) were analyzed independently by two reviewers, one of whom was blinded to treatment group. The blinded reviewer counted 8.0±5.3 seizures per recording, whereas the unblinded reviewer counted 8.3±4.6 seizures per recording. Paired analysis confirmed that seizure counts did not differ significantly between the blinded and unblinded reviews (p = 0.7728; Wilcoxon). The data suggest that blinding ECoG reviewers to treatment group in the present study did not confound seizure identification.
Tacrolimus did not protect against cortical atrophy or thalamic calcifications following TBI (Experiment 1)
Lateral fluid percussion TBI causes progressive degeneration of ipsilateral cortex over the course of a year[31], and the degeneration of certain cortical regions correlates with the incidence of late spontaneous seizures[26]. Therefore, to determine the location and extent of cortical degeneration in the present study, we sacrificed the rats of Experiment 1 at 43 ± 8 weeks post-TBI (or sham) and processed them for histology, along with age-matched controls. Groups did not differ significantly either in age at sacrifice or in time between TBI and sacrifice (age at sacrifice, F[2,20] = 1.438, p = 0.2610; time between TBI and sacrifice, F[2,14] = 0.1998, p = 0.8212; Table 1). Gross examination of fixed brains from TBI rats revealed an opaque lesion, 1mm or more in diameter, at or just ventral to the site of fluid percussion impact. We collected coronal sections at 0.6mm intervals between 0.8mm and 5.6mm caudal to Bregma, stained the sections with thionine and examined them by light microscopy (Figure 5A & 5B). Most TBI brains showed at least 1 opaquely-stained lesion in the ipsilateral thalamus (Figure 5C and inset in Figure 5B). We interpreted these lesions as thalamic calcifications, based on similar findings in previous studies of fluid percussion TBI[26,28,32].
Thalamic calcifications were identified in nearly all TBI rats but never in control rats (Figure 5C). However, the prevalence of these calcifications did not differ significantly between TBI Only and TBI+TAC rats (χ2[1] = 0.7804; p = 0.3770; Figure 5C). Nor did the presence of calcifications correlate with seizure frequency, either within or across groups (p>0.05). The data therefore suggest that thalamic calcifications were not related to the frequency of non-convulsive seizures after lateral TBI.
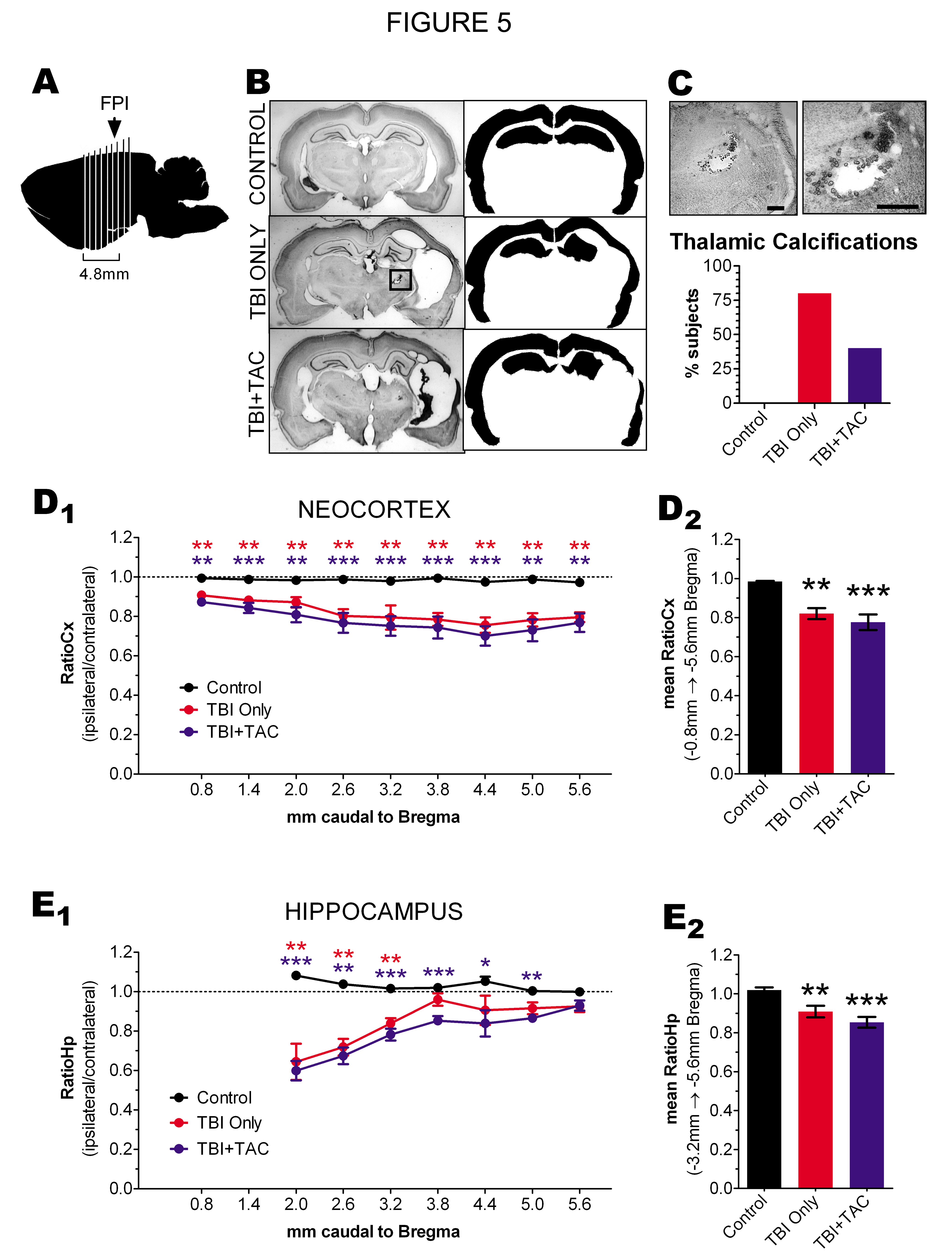
Figure 5:
Lateral TBI Caused Thalamic Calcifications and Atrophy of Ipsilateral Cortex: Rats were sacrificed and processed for thionine histology after completion of video-ECoG, approximately 42 ± 9 weeks after lateral fluid percussion TBI, or at an equivalent age in uninjured controls. A) Illustration indicating where sections were collected relative to the site of fluid percussion impact (FPI). B) Low-magnification (0.5x) light micrographs of representative, thionine-stained sections from -3.8mm Bregma, and the corresponding traces of neocortex and hippocampus which were used for measuring area (in black). Inset indicates a thalamic calcification; a smaller calcification can also be seen in the TBI+TAC image. C) Thalamic calcification from (B, inset) shown at 5x and 10x magnification, and a graph of the percentage of rats having at least 1 thalamic calcification. D) TBI Only and TBI+TAC rats showed significant but comparable decreases in RatioCx when assessed at each Bregma level (D1) and when averaged across Bregma levels (D2). E) TBI Only and TBI+TAC rats showed significant but comparable decreases in RatioCx when assessed at the rostral Bregma levels (E1) and when averaged from Bregma -3.2mm to -5.6mm (E2). The decrease in RatioHp in the rostral sections (Bregma -2.0mm and -2.6mm) appeared to be due to shrinkage of the hippocampus along the rostro-caudal axis; the affected sections were therefore excluded from the mean RatioHp. Means compared by Kruskal-Wallis test with post hoc Mann Whitney tests. Scale bar, 0.5mm. *p < 0.05 vs. control, **p < 0.01 vs. control, ***p < 0.001 vs. control
Initial examinations also suggested atrophy of the ipsilateral neocortex in all TBI rats. To quantify the apparent atrophy, we used ImageJ to trace the ipsilateral and contralateral neocortex in micrographs and then measure the area enclosed by the tracing (Figure 5B). The traced area of the ipsilateral (left) neocortex was then divided by that of the contralateral (right) neocortex in each brain section, yielding a ratio of the two hemispheres ("RatioCx")[26]. This analysis revealed significant differences in RatioCx among the treatment groups, as described below.
NEOCORTEX - The RatioCx in control brains was consistent along the rostro-caudal axis, averaging 0.98 ± 0.01 when measured from -0.8mm through -5.6mm Bregma (Figure 5D1). Cross-comparison of the mean RatioCx revealed significant differences among treatment groups (mean RatioCx, p = 0.0022; Figure 5 D2). Post hoc analysis indicated, for example, that the RatioCx was significantly decreased in TBI Only rats relative to controls (p = 0.0043). The RatioCx in TBI+TAC rats was also decreased from control levels (p = 0.0002). The decrease in RatioCx in TBI+TAC rats appeared to be comparable to that of TBI Only rats, as the TBI groups did not differ significantly from one another (p = 0.6787). RatioCx decreased most likely due to atrophy of the ipsilateral neocortex, since the area of the contralateral neocortex did not differ significantly across treatment groups (control, 10.6 ± 1.4 arbitrary units, A.U.; TBI Only, 9.9 ± 0.3 A.U.; TBI+TAC, 10.4 ± 0.9 A.U.; p = 0.3228, Kruskal - Wallis) (Kharatishvili and Pitkanen, 2010). The data thus demonstrate substantial atrophy of the ipsilateral neocortex by 43 weeks post-TBI, and agree with previous findings that Tacrolimus does not prevent neocortical atrophy following TBI[33,34].
Previous research has shown that the risk of spontaneous seizures increases with the severity of neocortical atrophy following lateral fluid percussion TBI[26]. We therefore used linear correlation to test whether seizure frequency and neocortical atrophy were related. However, mean RatioCx did not correlate with the frequency of non-convulsive seizures, either within TBI groups or when all TBI groups were combined (each, p>0.05). The data thus suggest that neocortical atrophy is not related to non-convulsive seizure frequency after lateral TBI.
We did not perform correlational analysis on convulsive seizure data due to lack of data. It is worth noting, however, that the two rats exhibiting convulsive seizures on video-ECoG also had the most severe neocortical atrophy (i.e., first and second lowest mean RatioCx among all rats). Quantitative analysis revealed that the RatioCx was significantly decreased in these two rats, when compared to the TBI rats in which no convulsive seizures were observed (t[13] = 4.563, p = 0.0005). The data therefore support previous research showing a correlation between neocortical atrophy and convulsive seizures after lateral TBI[26].
Hippocampus
We also assessed atrophy in the hippocampus, since previous research has linked hippocampal asymmetry related to the incidence of late non-convulsive seizures after fluid percussion TBI[29]. In the present study, we estimated hippocampal atrophy by tracing the ipsilateral and contralateral hippocampi, measuring the enclosed areas (Figure 5B), and then calculating a ratio of ipsilateral to contralateral area ("RatioHp"). Analysis of RatioHp was limited to Bregma levels -3.2mm through -5.6mm for two reasons. First, hippocampal gray matter was not present in the sections representing -0.8mm and -1.4mm Bregma. Second, the rostral end of the hippocampus appeared to have shifted caudally after TBI, confounding the RatioHp in those sections (Figure 5E1).
RatioHp in controls was consistent along the rostro-caudal axis, averaging 1.03 ± 0.03 when measured from Bregma levels -3.2mm to -5.6mm (Figure 5E1). As with RatioCx, RatioHp differed significantly among treatment groups (mean RatioHp, p = 0.0028; Figure 5E2). In particular, RatioHp in TBI Only rats was significantly decreased relative to controls (p = 0.0087). TBI+TAC rats also showed a loss of RatioHp when compared to controls (p = 0.0005), but not when compared to TBI Only rats (p = 0.3710). As with RatioCx, RatioHp most likely decreased due to changes in the ipsilateral hippocampus, since the area of the contralateral hippocampus did not differ significantly across treatment groups (control, 4.6 ± 0.4 arbitrary units, A.U.; TBI Only, 4.2 ± 0.4 A.U.; TBI+TAC, 4.6 ± 0.7 A.U.; p = 0.3646, Kruskal-Wallis). Overall, the data suggest that the ipsilateral hippocampus underwent a significant change in volume and/or shape by 43 weeks post-TBI. While this change is consistent with previous reports of hippocampal asymmetry after fluid percussion injury[29], it did not appear to be related to the expression of non-convulsive seizures, since RatioHp did not correlate with non-convulsive seizure frequency (within or across the TBI groups, p>0.05).
Seizures were not observed at 5 weeks post-tbi (Experiment 2)
Spontaneous non-convulsive seizures begin as early as a few weeks after lateral fluid percussion TBI, and increase in both frequency and severity over months[13]. To characterize the timing of seizure development in the present study, we subjected a different set of rats to lateral fluid percussion TBI and then monitored spontaneous seizure activity by video-ECoG 5 weeks later (Experiment 2; Figure 6A). The severity of TBI in Experiment 2 was equivalent to that in Experiment 1 (see Results above), and the treatment groups were also the same - rats received either a single dose of Tacrolimus (5mg/kg; i.p.; "TBI+TAC"; n = 4) or no injection ("TBI Only"; n = 5) at 1 hour post-TBI. At 5 weeks post-TBI, injured rats and their uninjured littermates (n = 4) were monitored by simultaneous video-ECoG. Video-ECoG was recorded 12±1 hours per rat, beginning in the morning and completed the same day. A trained observer who was blinded to treatment group then visually reviewed video-ECoG data offline for evidence of seizure activity.
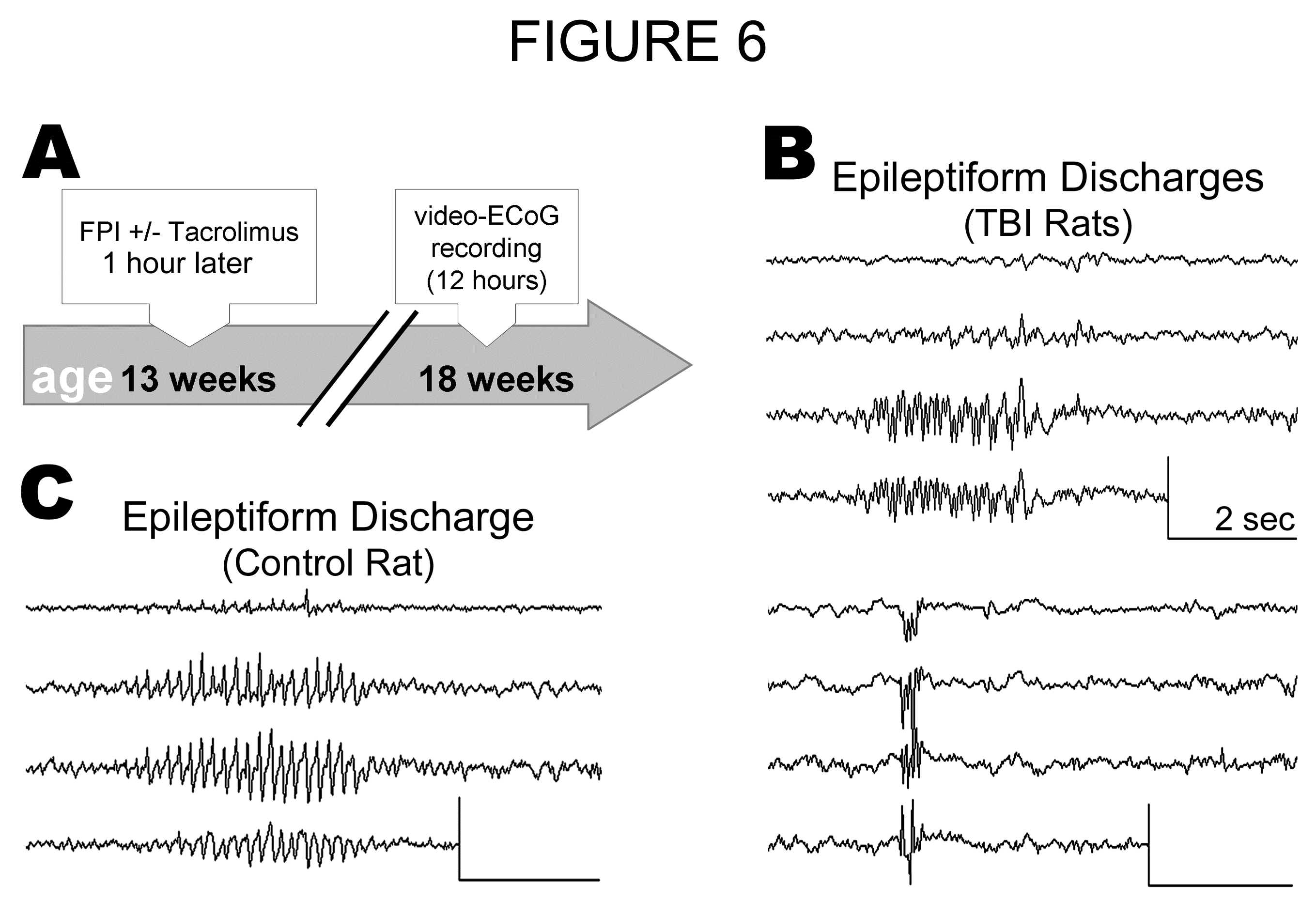
Figure 6:
No Seizures Were Observed at 5 Weeks Post-TBI: Adult, male Sprague-Dawley rats were subjected to lateral fluid percussion TBI, followed 1 hour later by administration of Tacrolimus (5mg/kg; i.p.; "TBI + TAC"; n = 4) or no injection ("TBI Only"; n = 5), and then were video-ECoG monitored at 5 weeks post-TBI along with uninjured littermates ("Control;" n = 4). A) Timeline of Experiment 2. B) Examples of paroxysmal electrographic discharges lasting < 5 seconds from two different TBI Only rats. Similar phenomena were observed occasionally (< 1/hr) in all TBI rats and were interpreted as epileptiform discharges. C) An example of the epileptiform activity exhibited by one control rat. All scale bars, 0.4mV by 3 seconds.
Low-voltage, arrhythmic electrographic activity was observed during awake behavior in all rats. In contrast to the baseline ECoG activity were observed paroxysmal discharges lasting less than 5 seconds. These epileptiform discharges were observed occasionally (>1/hr) in all TBI rats (Figure 6B) and in one control rat (Figure 6C). However, epileptiform activity lasting longer than 5 seconds was not observed in any rat. The data thus confirms previous reports that seizure prevalence and/or frequency increases over months following fluid percussion TBI[13,28,29].
Discussion
The present study describes several major findings related to the long-term electrographic and anatomical consequences of lateral TBI. For example, video-ECoG revealed two distinct types of spontaneous seizures in adult rats months (but not weeks) after TBI. These seizure types included convulsive seizures which were infrequent but lasted for minutes, and non-convulsive seizures which were frequent but lasted tens of seconds. Interestingly, administering a calcineurin inhibitor, Tacrolimus (TBI+TAC) at 1 hour post-TBI significantly decreased the frequency of non-convulsive seizures months later but did not protect against cortical atrophy or thalamic calcifications. The context and implications of these findings are discussed below.
Previous studies of similar TBI models have reported either convulsive seizures or non-convulsive seizures in the months following TBI[13,14]. For example, Kharatishvili and colleagues (2006) described infrequent but long-lasting convulsive seizures in up to 50% of rats after TBI, but did not note any non-convulsive seizures[14]. In contrast, a study by Curia et al. (2011) found frequent but short-lasting non-convulsive seizures in nearly all rats after TBI, but did not note any convulsive seizures[13]. The present study is thus the first (to our knowledge) to identify both seizure types in the same model, and in the same rats. Methodological differences could explain discrepancies among the present and previous studies. For example, the young rats (1 month old) used in the Curia et al. (2011) study may be less prone to develop convulsive seizures after TBI than the adult rats used in the present study and in the Kharatishvili et al. (2006). Age-dependency is characteristic of other models of acquired epilepsy – for example, rats younger than 25 days are less likely to develop spontaneous convulsive seizures following experimental status epilepticus[35,36].
Studies of post-traumatic epilepsy in human patients and rodent models suggest that epileptogenesis begins within minutes to hours of TBI[6,7], though the mechanism remains unknown. Our previous research identified an increase calcineurin activity in the minutes to hours following lateral fluid percussion TBI[15]. This increase in calcineurin activity could be an early but critical event in the development of post-traumatic epilepsy, as has been proposed in other models of acquired epilepsy (reviewed in[9]). Consistent with this possibility, the present study demonstrated that administering a calcineurin inhibitor, Tacrolimus, 1 hour after TBI reduced the frequency of spontaneous non-convulsive seizures months later.
Add Dose Selection Here
This finding may shed light on the timing and mechanisms by which non-convulsive seizures develop after TBI, as it reveals a Tacrolimus-sensitive mechanism which is engaged acutely after TBI. Though only a single dose of Tacrolimus was administered in the present study, previous studies suggest that brain levels can remain stable for days[37]. In addition, the dose used in the present study has been shown to inhibit calcineurin activity as measured by DARRP-32 dephosphorylation, cofilin dephosphorylation and cofilin-actin binding (Kurz et al., 2005). It is therefore possible that Tacrolimus inhibited calcineurin activity for days after TBI, potentially blocking the increase in calcineurin activity our lab previously identified in this model[15]. Further work is needed to characterize calcineurin inhibition by Tacrolimus in this TBI model, and to confirm that calcineurin inhibition is sufficient for disease-modifying effects. Regardless, the results of the present study suggest that pharmacological intervention at a clinically-relevant time point can alter the course of epilepsy development after TBI.
Further research is also needed to understand how calcineurin could mediate epileptogenesis. One possibility is that a pathological increase in calcineurin activity leads to the dysregulation of hyperpolarization-activated cation (HCN) channels[10]. Calcineurin has been shown to down regulate gating of HCN channels, resulting in neuronal hyper excitability[10], and a reduction in HCN channel expression in rat cortex is associated with epileptogenesis after status epilepticus[38,39]. HCN channelopathy may also explain the spontaneous non-convulsive seizures observed in the present study. HCN channels are down regulated in subthalamic nuclei in the tottering mouse, a genetic model of spontaneous non-convulsive seizures[40]. In addition, mice lacking a subtype of HCN channels (HCN2) also exhibit spontaneous non-convulsive seizures[41]. It is therefore possible that TBI induces (or exacerbates) the development of spontaneous non-convulsive seizures through a calcineurin-dependent dysregulation of HCN channels, and that this process begins within hours or days of TBI.
Calcineurin may also mediate epileptogenesis through its effects on the immune system or in synaptic circuits. For example, calcineurin mediates pro-inflmmatory signaling by interleukin-1β and tumor necrosis factor-α, and these signaling pathways have been strongly implicated in epileptogenesis[42 - 45]. An alternative but not exclusive possibility is that calcineurin facilitates epileptogenesis by destabilizing dendritic spines. Various injury models have shown that drugs which inhibit calcineurin activity also block the loss of dendritic spines after injury, including the spine loss we observed in the neocortex and dentate gyrus after lateral TBI[17 - 20]. Since dendritic spines bear the majority of excitatory synapses in the adult cortex[46], a widespread and persistent loss of spines could equate to deafferentation. Deafferentation can induce epileptiform activity in the neocortex[47-49], potentially through a homeostatic increase in network excitability[50]. In addition, studies of human epileptic tissue have found that spine loss in the dentate gyrus correlates with an abnormal prolonging of excitatory post-synaptic responses[51-53], which could support seizure activity. Further work is needed to understand whether and how dendritic spine loss, as well as HCN channel dysregulation and inflmmation, contribute to epileptogenesis. Yet, considering the variety of potential mechanisms, calcineurin may prove to be a valuable target for ameliorating post-traumatic epilepsy.
Acknowledgment
The authors gratefully acknowledge Drs. Robert Hamm and Tom Reeves for use of the fluid percussion injury devices. This study was funded by an Alzheimer's and Related Diseases Research Award Fund (Award Number 11-1, to S.B.C.; www.sahp.vcu.edu/vcoa) and a National Research Service Award Institutional Research Training Grant (5T32NS007288-25; www.nih.gov). Microscopy was performed at the VCU Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, with funding from NIH-NINDS Center core grant (5P30NS047463-02).










