
Using Integrated Bioinformatics Strategy to Identify Critical Factors for the Structural Integrity of Salmonella T3SS
Seth Ingram1, Bo He2, Paige DePagter1, Bin Xue1*
Affiliation
1Department of Cell Biology, Microbiology and Molecular Biology, School of Natural Sciences and Mathematics, College of Arts and Sciences, University of South Florida, Tampa, FL 33612, USA
2College of Automation, Harbin Engineering University, Harbin, Heilongjiang 15001, China
Corresponding Author
Bin Xue, Department of Cell Biology, Microbiology, and Molecular Biology, College of Arts and Sciences, University of South Florida, 4202 E. Fowler Ave, ISA 2015, Tampa, FL, USA 33620, Tel: (813) 974-6007; E-mail: binxue@usf.edu
Citation
Xue, B., et al. Using Integrated Bioinformatics Strategy to Identify Critical Factors for the Structural Integrity of Salmonella T3SS (2016) Bioinfo Proteom Img Anal 2(1): 76- 84.
Copy rights
Keywords
Protein intrinsic disorder, MoRF, Binding motif
Abstract
Type-III secretion system of Gram-negative bacteria is the major molecular machine responsible for the infection of host cells and the in-host survival of the bacteria. The T3SS is composed of three structural components: basal body, needle, and export apparatus. The needle is an extracellular protein complex that recognizes host cells and transport bacterial effector proteins into the host cells. The basal body forms a channel across the bacterial membranes and also provides structural support to the needle. The export apparatus selects effector proteins and initiates the transportation of these proteins. Since all these three structural components are formed by specific proteins, abolishing the interaction of these proteins will disrupt the structural integrity of one or more structural components of T3SS, and eventually affect the infection and/or virulence of the bacteria. In this study, we analyzed the sequential, structural, and interactomic features of Salmonella T3SS structural proteins. We found that these structural proteins have abundant short and/or long disordered regions that overlap with other structured/functional regions. We identified critical interaction patterns and hub proteins SipB, SpaO, and SpaS, in the interactome of T3SS structural proteins. We also predicted novel binding motifs for six T3SS structural proteins of which the interaction partners are unknown. These results are expected to shed light on future studies in the fields of T3SS structural integrity and drug discovery.
Introduction
Type III Secretion System (T3SS) is a needle-like appendage found on the surfaces of many gram-negative bacteria, such as: Salmonella, Escherichia coli (E.coli), Vibrio, Yersinia, and Chlamyida. T3SS facilitates these bacteria to invade host cells and to cause various diseases, including: typhoid fever, food poisoning, diarrhea, plague, and many others[1,2]. A T3SS apparatus measures 60~80 nm in length and 8 nm in diameter with an interior lumen of ~3 nm in diameter. The major function of T3SS is to export specific proteins (effector proteins) from bacterial cytoplasm to the extracellular to invade and to manipulate host cells. For this reason, T3SS is also called injectisome or injectosome. A single bacterium normally expresses one but may have more T3SSs[3,4]. The bacterial genome may contain different clusters of genes that express different types of T3SS. Salmonella contains two clusters of T3SS genes, which are Salmonella Pathogenicity Island 1 (SPI-1) that is responsible for host invasion, and Salmonella Pathogenicity Island 2 (SPI-2) that is critical for the survival of the bacterium inside host cell[5]. These two systems are similar to each other, but also present remarkable differences[6]. Enterohaemorrhagic E. coli also has two clusters of T3SS genes[7,8]. However, thefunctional differences between them are not completely discovered.
T3SSs from various bacterial species share very similar structures. A T3SS appendage contains three structural parts: transmembrane basal body, extracellular needle, and cytoplasmic export apparatus[9]. The structures of the basal body and needle are similar to that of the base and hook of flagellar filament[10], indicating their common origin of evolution. The basal body is integrated with two layers of membrane of bacterium and is thus composed of three portions: a ring integrated with the outer membrane of the bacterium (OM ring), a ring integrated into the inner member of the bacterium (IM ring), and a periplasmic rod connecting both rings. The needle is connected to the basal body and extends to the extracellular part of the bacterium. The needle is a syringe-like structure composed of needle filament, tip, and translocon. The needle filament is composed of many proteins of the same type, which enclose an interior lumen with a diameter of 3 nm to facilitate the secretion of effector proteins[11-15]. The tip of T3SS needle is able to trigger the secretion of effector proteins[13] upon being activated by specific environmental factors, such as contact with specific host cells, temperature, pH value, osmolarity, and many others[16]. The export apparatus is a dynamic complex of multiple proteins connected to the IM ring of T3SS[17,19]. The function of the export apparatus is to control the secretion of effector proteins. After being secreted into host cells, these effector proteins are able to manipulate the host cells in multiple ways[20-23]. More specific examples of host cell manipulation include: inducing the host cell to engulf the bacterium; inducing apoptosis[24] by the interaction between Shigella flexneri effector IpaB and caspase 1 of host cell[25]; and entering the nucleus of host cell and then activating the expression of genes that are beneficial for bacterial infection as shown in thecase of Xanthomonas effector TAL[26]. Deletion of proteins in the export apparatus significantly influenced the secretion of other proteins[17,19,27-30], and hence may alter the virulence and ability of affection of the bacteria.
Clearly, T3SS is critical for the pathogenicity of Gram-negative bacteria. Recent studies found that defects of T3SS significantly reduced the ability of infection of bacteria[31-35]. Therefore, disrupting the assembly of T3SS eliminated the virulence, but didn’t kill the bacteria. These observations suggested a new alternative strategy for drug development by focusing on anti-virulence and anti-infective targets[37-40]. This alternative strategy does not add selection pressure towards drug resistance and hence may indicate a solution to the fast developed drug resistance of traditional antibiotics that are normally either bactericidal or bacteriostatic[41], and provide additional clinical treatment of bacterial infection. For these reasons, T3SS has become a very important target for drug development[37]. Several small molecules have been reported to disrupt the assembly and function of T3SS in various models systems[42-44].
To facilitate mechanistic studies on the structural integrity of T3SS and to facilitate the discovery of new drugs that target T3SS structural proteins, thorough understanding on the three-dimensional structures and on the interaction partners of T3SS structural proteins becomes a prerequisite. The T3SS system is normally composed of tens of proteins, which are categorized into two groups: structural proteins that are responsible for the assembly of the basal body and needle of T3SS; and translocator proteins that facilitate the translocation of other bacterial proteins from bacterial cytoplasm to the outside. Many proteins that compose of T3SS are multi-domain proteins. 3D-dimentional structures for some of them have been identified[44-46]. However, not all the T3SS proteins are structured. Intrinsicallydisordered regions have been found in several individual T3SS proteins, such as Salmonella SipD[47], Ecoli EspD[48], Yersinia LcrV[49], and Pseudomonas PopB[50]. These disordered regions are frequently involved in protein-protein interactions[47,48,50-52]. Nonetheless, the overall abundance and functional importance of intrinsic disorder in the T3SS proteins still remains unclear.
In our previous analysis on the distribution of proteinintrinsic disorder among over 3,500 species, bacterial proteomes were estimated to have about 20-24% of intrinsic disorder each[53]. In many pathogens, disordered regions are correlated tothe virulence[54-60]. Therefore, it becomes interesting to see how much intrinsic disorder is in the T3SS system and what functions it has. In this study, we characterized various sequential, structural, and functional features of the structural proteins of Salmonella T3SS, identified their interaction patterns, and predicted novel binding motifs. The results of the study can be used for further studies of identifying new targets and developing new strategies to disrupt the assembly of T3SS and therefore to reduce the infection and virulence of bacteria.
Methods
T3SS structural proteins Since the proteins that make up T3SS are highly conserved across many different species of Gram-negative bacteria on their sequences, structures, and functions[61-63], we chose T3SS structural proteins of Salmonella typhimurium SPI-1 in this study. The number of T3SS structural proteins in Salmonella was estimated to be around 20 in previous studies[64]. Among which, several proteins may have general-purpose functions. Therefore, we selected nineteen proteins as listed in Table 1. These nineteen proteins may be split into three groups: (1) Basal body structural proteins: PrgH, PrgK, InvG, InvH, and PrgJ; (2) Needle structural proteins: PrgI, SipD, SipB, and SipC; and (3) Export apparatus structural proteins: InvC, InvI, OrgB, SpaO, InvA, InvE, SpaP, SpaR, SpaQ, and SpaS. The amino acid sequences of these proteins were extracted from UniProt. The 3D structures of these proteins or of segments of these proteins were extracted from PDB.
Protein-protein interaction network Both STRING[64] and DIP[65] databases were used to search for interaction partners of the T3SS structural proteins. STRING is one of the most well maintained and most prevailing databases for protein-protein interactions. We only selected interactions that had been experimentally validated for our further analysis. DIP is another well designed database for experimentally identified protein-protein interactions. The information from DIP was used to supplement the results of STRING.
Table 1: List of Salmonella T3SS structural proteins.
| Structural parts | Protein Name | Length | Function | UniProt ID | PDB ID | PDB Sequence | PDB Coverage |
| Basal body | PrgH* | 392 | IM ring | P41783 | 4G2S; 4G1I | 11-119; 170-392 | 84.7% |
| PrgK* | 252 | IM ring | P41786 | 4W4M; 4OYC | 19-92; 96-200 | 73.8% | |
| InvG | 562 | OM ring | P35672 | 4G08 | 22-178 | 27.9% | |
| InvH | 147 | OM ring | P0CL43 | --- | --- | --- | |
| PrgJ | 101 | Inner rod | P41785 | --- | --- | --- | |
| Needle | PrgI | 80 | Filament | P41784 | 3ZQE | 1-80 | 100% |
| SipD | 343 | Tip | Q56026 | 3NZZ | 38-110; 132-342 | 82.8% | |
| SipB** | 593 | Translocon | Q56019 | 3TUL | 83-115; 127-170; 182-226 | 20.6% | |
| SipC** | 409 | Translocon | P0CL47 | --- | --- | --- | |
| Export apparatus | InvC | 431 | ATPase | P0A1B9 | --- | --- | --- |
| InvI (SpaM) | 147 | Central stalk | P40612 | --- | --- | --- | |
| OrgB | 226 | Peripheral stalk | E1WAB4 | --- | --- | --- | |
| SpaO | 303 | C-ring homolog | P40699 | 4YXA | 145-213; 232-297 | 44.6% | |
| InvA** | 685 | Gate | P0A1I3 | 2X49 | 357-685 | 47.9% | |
| InvE | 372 | Gate-keeper | P35671 | --- | --- | --- | |
| SpaP** | 224 | IM partner | P40700 | --- | --- | --- | |
| SpaR** | 263 | IM partner | P40701 | --- | --- | --- | |
| SpaQ** | 86 | IM partner | P0A1L7 | --- | --- | --- | |
| SpaS** | 356 | P40702 | 3C01 | 239-345 | 30.0% |
N.B. (*) Proteins with single trans-membrane segment. (**) Proteins with multiple trans-membrane segments. When proteins have multiple PDB entries, the X-ray structure with firstly the highest sequence coverage and then the highest resolution was selected. The PDB ID column may have more than one PDB IDs when the protein has multiple PDB entries for different segments. A single PDB ID may contain multiple segments of the same protein as shown by the numbers in the PDB Sequence column. In this case, the missing residues between segments were not crystallized and are hence disordered. The values in the PDB Coverage column present the fractions of amino acids included in the PDB structures.
Disorder prediction
PONDR-VLXT[66] and PONDR-FIT[67] were used to predict the structural flexibility of proteins. PONDR-VLXT is very powerful in identifying hydrophobic clusters inside intrinsically disordered regions. PONDR-FIT is one of most accurate predictors for disorder prediction and for providing biologically relevant information[68]. The combination of PONDR-VLXT and PONDR-FIT was used by us to analyze the correlation between protein intrinsic disorder and function in many projects, such as reprogramming factors of induced ploripotent stem cells[69], structural flexibility and mechanisms for methionine oxidation[70], evolution of P53[71], PTEN interactome[72], virulence factors[73], yeast mitosis factors[74], DBC1[75], Emerin[76], etc. Both of these two predictors take amino acid sequence as input. The output is a list of per-residue scores for all the residues in the sequence. Residues with scores higher than 0.5 are assigned as disordered residues, while residues with scores lower than 0.5 are interpreted as structured residues. Consecutive disordered residues form an Intrinsically Disordered Region (IDR). When all the residues in a sequence are disordered, the entire protein is predicted to be Intrinsically Disordered Protein (IDP).
Secondary structure prediction
NetSurfP[77] was used to predict the secondary structure of protein sequences. NetSurfP is an ensemble predictor that provides not only the high-quality predictions, but also the reliability of each prediction. In addition to secondary structure, NetSurfP also provides prediction of accessible surface area.
Functional motifs/domains
Pfam[78] was used to search for functional domains of the T3SS structural proteins. We also used MoRF-II[79,80] and ANCHOR[81] to predict potential binding motifs inside each of the T3SS structural proteins. MoRF-II was designed to identify short motifs that locate inside intrinsically disordered regions and transform their conformations from coil to helix upon binding to partners. The binding motifs identified by MoRF-II very frequently overlap with dips in the disorder profile made from disorder scores predicted by PONDR-VLXT. ANCHOR is able to predict highly hydrophobic segments inside disordered regions based on the calculation of interaction energy. These two predictors are able to identify binding motifs of different preferences.
Results
As shown in Table 1, ten out of nineteen T3SS structural proteins don’t have PDB structures. In the rest nine proteins that have PDB structures, four have structures for at least 70% of each of the sequences, and the other five only have structures for less than 50% of each of the full length sequences. Therefore, most of the T3SS structural proteins or protein regions still do not have experimentally validated structures. To check whether or not those structure-unknown proteins and/or regions are IDPs or IDRs, PONDR-FIT was used to predict all the nineteen proteins. The per-residue predictions for all the proteins were presented in Figure 1(a). Curves below dashed lines represent structured regions, of which the 3D structures should be able to obtain through experimental methods. Curves above dashed lines denote IDRs that don’t have rigid 3D structures under physiological conditions. When analyzing the results, the predictions of structure-known proteins and regions were used as a control set to compare with their corresponding experimentally observed structured regions to examine the prediction accuracy of the predictor. Clearly, all the regions that have PDB X-ray structures have been predicted to be structured except ~AA160~240 of SipB and ~AA40~110 of SipD. These two regions possess structures of helical bundle as shown in Figure 1(b). The amino acid sequences corresponding to these two regions are highly charged and hydrophilic. Nonetheless, segments of these two sequences form amphiphilic helices as demonstrated in the figure by the colors on different sides of these helices. These amphiphilic helices use their hydrophobic sides to interact with each other and to form helical bundles. After taking this factor into consideration, it is clear that the results of PONDR-FIT prediction match to the experimental results very well.
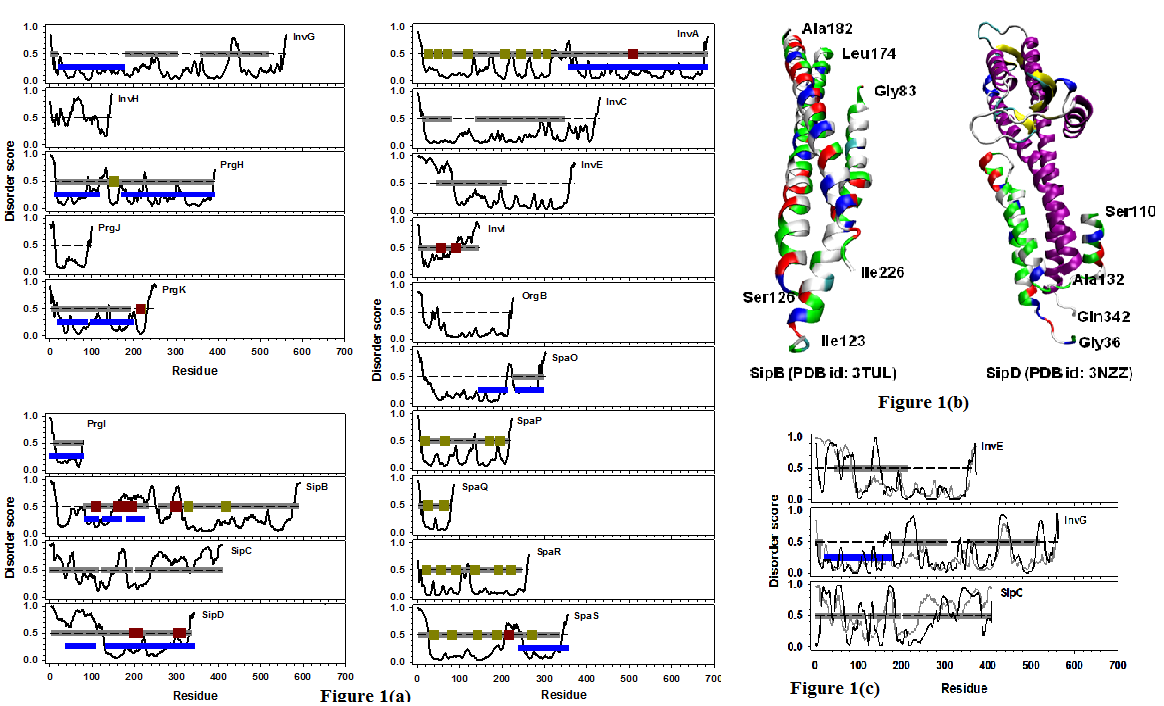
Figure 1: (a) Disorder prediction of nineteen Salmonella T3SS structural proteins. The proteins were organized in three groups: proteins in basal body (upper left panel), proteins in needle (lower left panel), and proteins in export apparatus (right panel). The name of each protein was labeled in the corresponding plot. In all the plots, x-axis is the index of amino acid along the sequence, and y-axis shows the predicted per-residue disorder score generated from PONDR-FIT. The curve in each plot is the disorder profile made from PONDR-FIT predictions for all residues in each protein. Dashed line in the middle of each plot indicates the boundary between disordered (y > = 0.5) and structured (y < 0.5) residues. Horizontal bars are categorized by their colors: blue - regions that have PDB structure; gray - Pfam domains; dark green – transmembrane segments; dark red – coiled coils. (b) PDB structures of two needle structural proteins: SipD (PDB id: 3NZZ) and SipB (PDB id: 3TUL). The region from Ala132 to Gln342 of SipD was colored by its secondary structures. The other region of SipD (Gly36-Ser110) was colored by the types of amino acids (white - hydrophobic; red – negatively charged; blue – positively charged; and green - polar). SipB was also colored by the types of amino acids in the same way. In both structures, discontinued regions indicate missing residues in the structure. (c) Combined analysis of disorder predictions from both PONDR-FIT (gray) and PONDR-VLXT (black) for InvE, InvG, and SipC. This plot is amended from (a) and therefore all the other annotations are the same as those in (a).
Disorder prediction also identified other structured regions that don’t have experimentally observed structures. These predicted structured regions can be classified into four groups: (1) Regions with multiple transmembrane segments, such as ~AA20-320 of InvA, ~AA10-210 of SpaP, ~AA10-80 of SpaQ, ~AA10-260 of SpaR, ~AA20-200 of SpaS, and ~AA320-420 of SipB. It has been well realized that solving the structure of transmembrane proteins is really challenging; (2) Regions that are shorter than 50~60 residues, e.g. ~AA180-AA240 of SipC. Many other proteins also have such short structure-prone regions. These short structure-prone regions may not have strong enough hydrophobic interactions to maintain rigid 3D structures[76]; (3) Regions overlapped with Pfam domains, including: ~AA180~320 and ~AA360-420 of InvG, ~AA20-80 and ~AA140-340 of InvC, ~AA40-210 of InvE, and SipC. Further analysis on disorder prediction of both PONDR-FIT and PONDR-VLXT as shown in Figure 1(c) indicated that each of these Pfam domains is a combination of short structural-prone region(s) and long IDR(s). For this reason, although the functional roles of these regions have been characterized, the structures of these domains are still not defined; (4) regions with uncharacterized features and functions, such as ~AA10-AA100 of PrgJ, AA220-360 of InvE, ~AA20-140 of SpaO, and ~AA60-320 of OrgB.
The predicted protein intrinsic disorder is not neglectible in the T3SS structural proteins, with 21.2% of disordered residues by PONDR-FIT predictor and 27.3% of disordered residues by PONDR-VLXT predictor. Almost all the proteins have disordered residues at N- and/or C-termini. Disordered regions were also found in the middle of or throughout sequences. InvH and InvI are both disorder-dominant proteins. Although both of them have structure-prone regions, these regions are shortand have considerable levels of flexibility as indicated by the values of their disordered scores. Therefore, these two proteins may not form rigid structures under physiological conditions. Another five proteins (SipB, SipC, SipD, InvE, and OrgB) have long IDRs that have at least 30 consecutive disordered residues. In which, the IDRs of SipB and SpaS connect other structured/functional domains, and the IDRs in the rest three proteins form entire N- or C-terminal disordered domains. In addition to long IDRs, short IDRs can be observed in all other proteins, such as inside functional Pfam domains (~AA420-460 of InvG), linking different structured domains (~AA210-230 of SpaO), separating transmembrane segments (~AA100-130 of SpaR), and inside structured domains (~AA220-230 of PrgH).
Figure 2 presents analysis on the abundance of intrinsic disorder in these proteins. Each protein in this figure is described by three bars from left to right representing: length of the protein, length of the longest IDR in that protein, and fraction of disordered residues in that protein. The first quantity shows the dimension of the protein. The other two quantities describe different aspects of disordered content: length of longest IDR shows the dimension of a consecutive segment of disordered residues; fraction of disordered residues shows the total amount of disordered residues in the protein. These two quantities can be combined to characterize the distribution of disordered residues in a protein. For example, PrgI has 16.3% of disordered residues and its longest IDR has 12 residues. By taking into consideration that PrgI has 80 residues in total, it can be concluded that almost all of the disordered residues in PrGI are in the longest disordered region. Another example is SipC, which contains 57.5% of disordered residues and has 90 residues in the longest IDR. Since this protein has 409 residues, it can be expected that this protein may have multiple long IDRs. When measuring the overall abundance of protein intrinsic disorder, seven proteins (InvH, SipD, SipB, SipC, InvI, InvE, and SpaP) out of nineteen contain IDRs longer than 30AA, these seven proteins also have more than 25% of disordered residues in their sequences. In which, SipC has the highest fraction of disordered residue of 57.5%. InvI, InvH, and SipD have more than 40% of disordered residues. In terms of the length of IDR, SipD has the longest IDR of 126 residues. SipB, SipB, and InvE have long IDRs that are near or over 90 residues. In addition, SpaP, InvI, and InvH have long IDRs that have at least 30 disordered residues.
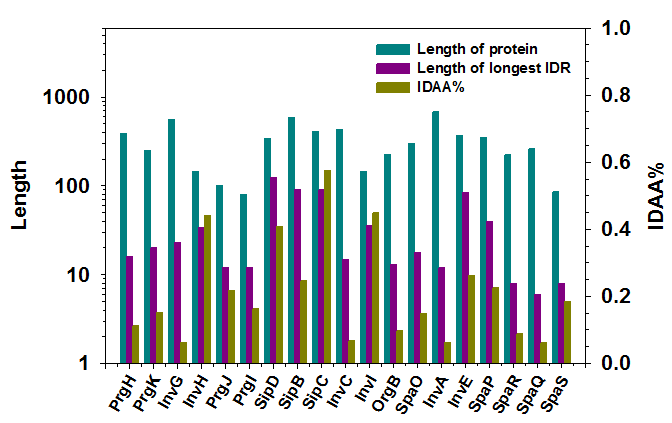
Figure 2: Distribution of predicted protein intrinsic disorder in nineteen structural proteins of Salmonella T3SS. X-axis shows 19 proteins listed in Table 1. Two y-axes were used in the figure. The first y-axis is on the left and shows the number of amino acids in either each protein (protein length, dark cyan) or the longest IDR (dark pink). The second y-axis is on the right and presents the fraction of predicted disordered residues in each protein (dark yellow). The long-dashed line corresponds to the 1st Y-axis and equals to 30AA. The short dashed line matches to 25% on the 2nd Y-axis.
Figure 3 shows protein-protein interaction networks of 19 T3SS structural proteins. Out of these 19 proteins, three proteins (SipB, SpaO, and SpaS) have interactions with proteins that do not belong to T3SS, ten (PrgI, PrgH, PrgJ, PrgK, InvC, InvG, InvA, SpaP, SpaQ, and SpaR) are bound by one or more T3SS structural proteins, the other six (SipC, SipD, InvH, InvE, InvI, and OrgB) do not have any experimentally validated interaction partners. In more details, all of the three proteins in the first group (SipB, SpaO, and SpaS) have multiple interaction partners. Both SipB and SpaS are transmembrane proteins with at least two transmembrane segments (Figure 1(a)). Meanwhile, both of them contain coiled coil(s) (Figure 1(a)). In the second group of which each of the ten proteins interacts with other T3SS proteins, three of them (PrgI, InvG, and SpaR) may each interact with itself. These three proteins either have PDB structures (PrgI, and N-terminal part of InvG), or were predicted to be structured (C-terminal of InvG, and SpaR). In addition, nine proteins (PrgI, InvG, PrgH, PrgJ, PrgK, InvA, SpaP, SpaQ, and SpaR) in the second group interact with SpaS. InvC, another protein in the second group, interacts with SpaO. SpaR also interacts with SpaP and SpaQ. Both SpaO and SpaS have another common interacting protein FliG, which is not a T3SS structural protein. In the third group that contains six non-interactive proteins, SipC and SipD are needle structural proteins, InvH is basal body structural protein located in the OM ring, the other three proteins (InvE, InvI, and OrgB) are components of export apparatus. In terms of functions, InvH and OrgB don’t have clear functional annotations, and the other four proteins may have PDB structure, Pfam domain, coiled coil, and/or transmembrane segment.
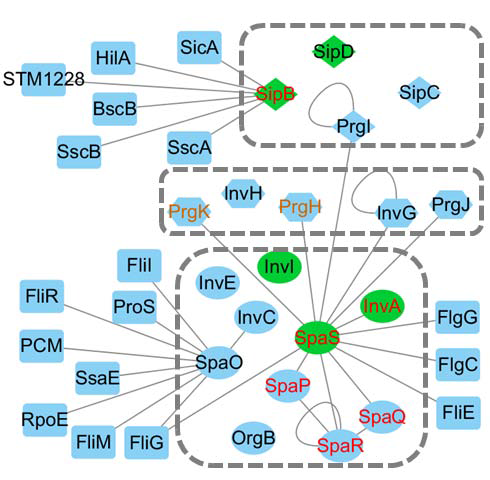
Figure 3: Protein-protein interaction networks for nineteen Salmonella T3SS structural proteins. Only physical interactions were considered in this figure. Each protein is a node and the edge between two nodes indicates that these two proteins have direct interaction. The nineteen T3SS proteins were organized into three dashed boxes from top to bottom, corresponding to needle structural proteins (diamond), basal body structural proteins (hexagon), and export apparatus proteins (eclipse). Nodes in green are proteins having coiled coils. Nodes with red labels are proteins with multiple transmembrane segments. Nodes with orange labels (only PrgH and PrgK) are proteins with single transmembrane segment. All other proteins in rectangles are non-T3SS Salmonella proteins that regulate T3SS structural proteins
To further explore the functional roles of the T3SS structural proteins, we predicted potential binding motifs of these proteins using both MoRF-II and ANCHOR predictors. Eight out of nineteen T3SS structural proteins were found to have predicted binding motifs as shown in Figure 4. InvH is one of the structural proteins in basal body. This protein is predicted to be highly flexible with several very short structure-prone segments. It doesn’t have any known structural or functional domains. Both of MoRF-II and ANCHOR identified a binding motif located at ~AA60 and ~AA80, respectively. Secondary structure analysis by NetSurfP showed that these two segments are helices. SipB, SipC, and SipD are needle structural proteins. SipB has multiple predicted binding motifs on/near both ends of its structure-known domain in the N-terminal half of the entire sequence. Since the structured domain is composed of coiled coils, the predicted binding motifs also intersperse on the linkers of those coiled coil segments. SipC has a ~60AA N-terminal disordered region, a ~200AA structure-prone domain in the middle, and a ~120AA disordered region at the C-terminal. All the predicted binding motifs locate in the C-terminal region. SipD has a ~40AA N-terminal disordered region, followed by another ~60AA helical bundle, and another ~200AA structured domain at the C-terminal. All the predicted binding motifs are in the N-terminal disordered region and/or the ends of helices in the helical bundle. In the export apparatus proteins, four (InvA, InvE, InvI, and OrgB) have predicted binding motifs. InvA has eight transmembrane segments in the N-terminal half and another structured domain in the C-terminal half. A predicted MoRF motif is right between the transmembrane domain and structured domain. InvE has an N-ter IDR followed by a C-ter structure-prone domain, with an identified Pfam domain covering the second half of the N-ter IDR and the first half of the C-ter structured domain. Predicted binding motifs are in the N-ter of the entire sequence or the N-ter of the Pfam domain. The locations of the predicted binding motifs from both MoRF-II and ANCHOR are consistent to each other. InvI is a short but almost fully disordered protein, with entire sequence being annotated as a Pfam functional domain. This domain has two coiled coils in the middle. Multiple binding motifs were predicted throughout the entire sequence. OrgB is another unannotated protein. It is composed of ~50AA N-ter IDR and a ~180AA C-ter structure- prone domain. A MoRF motif was identified in the N-ter IDR. In brief, T3SS structural proteins have multiple short binding motifs, which can be used to regulate the interaction between them and other proteins. More interestingly, all the six proteins that don’t have interaction partners in protein-protein interaction databases (Figure 3) were predicted to have binding motifs.
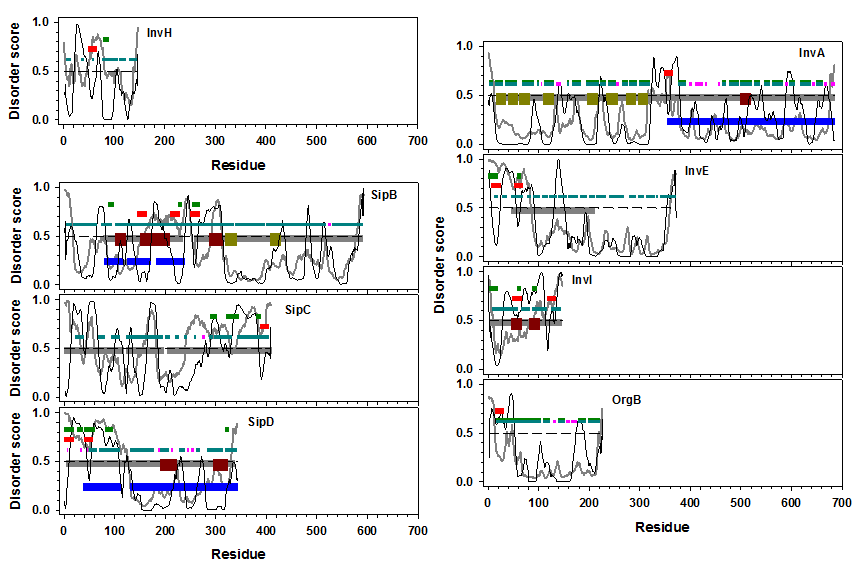
Figure 4: Predicted binding motifs in Salmonella T3SS structural proteins. Eight of nineteen proteins showed binding motifs predicted by MoRF-II and ANCHOR predictors and were therefore shown in this figure. These eight proteins are: (1) InvH in the basal body (upper left panel); (2) SipB, SipC, and SipD in the needle (Lower left panel); and (3) InvE, InvI, OrgB, and InvA in the export apparatus (right panel). In all these plots, x-axis shows the index of amino acid along the protein sequence, y-axis presents per-residue disorder scores predicted from both PONDR-FIT and PONDR-VLXT. Curves in gray are disorder profiles from PONDR-FIT prediction, while curves in black are disorder profiles from PONDR-VLXT prediction. The dashed lines in the middle are the boundary of disordered (y > = 0.5) and structured (y < 0.5) residues. Horizontal bars represent regions of specific interests (from top to bottom): dark green – binding motifs predicted by ANCHOR; red – binding motifs predicted by MoRF-II; dark cyan – helical segments predicted by NetSurfP; pink – beta-strands predicted by NetSurfP; gray – Pfam domains; dark red – coiled coils; dark yellow – transmembrane segments; blue – regions with PDB structures.
Discussion
T3SS is the major molecular machine responsible for bacterial infection and virulence of Gram-negative bacteria. The T3SS is composed of about 20 proteins by forming three structural sections: basal body, needle, and the export apparatus[45,46]. The needle is responsible for host detection and transport of infection and virulence factors. The basal body constructs a channel across bacterial membranes and provides structural support to the needle. The export apparatus facilitate the selection and transport initiation of various effector proteins. Each of the structural sections is formed by multiple proteins, which are also called T3SS structural proteins. Clearly, interrupting the interaction of structural proteins inside TS33 or manipulating the interaction between T3SS structural proteins and other bacterial proteins may disrupt the structural integrity of T3SS, and thus affect the affection and virulence of the bacteria. Such a strategy provides an alternative way for drug development that is not related to bactericidal or bacteriostatic and hence poses less selective pressure of developing drug resistance on the bacteria. Studies on the structural biology of the T3SS proteins are critical for understanding the structural integrity of T3SS and the manipulation of the T3SS structures.
Not all of the T3SS structural proteins have experimentally observed structures. By analyzing the results of disorder prediction from both PONDR-FIT and PONDR-VLXT predictors, we found that the T3SS structural proteins have significant amount of protein intrinsic disorder. The disordered residues stay in both N- and C-termini of each protein, connects various structured and/or functional domains, and form large disordered functional domains that may have more than one hundred residues. These disordered regions may contain various structural motifs, such as coiled coil, helix, and bets-strand, and are critical for intra- and intermolecular interaction[51].
By analyzing the protein-protein interaction networks of all the T3SS structural proteins, we identified several hub proteins in the networks and specific patterns of interaction. Among nineteen T3SS structural proteins, three (SipB, SpaO, and SpaS) have multiple non-T3SS interaction partners. SpaO and SpaS can both interact with FliG, a non-T3SS protein. SpaP, SpaQ, SpaR, and SpaS also form a closed loop in the protein-protein interaction network. PrgI, InvG, and SpaR are each able to form multimer with itself. Therefore, these results provide a new strategy of selecting critical target for regulating the interactions and assembly of T3SS.
Six out of nineteen T3SS structural proteins don’t have experimentally observed interaction partners. We applied MoRF-II and ANCHOR predictors and identified multiple binding motifs in these six proteins, as well as in another two proteins. The binding motifs are in terminal regions of proteins, in the linker region of structured domains, at the edge of structured domains, or inside long disordered regions. The discovery of these binding motifs provides critical information for the experimental validation of the interaction partners and interaction patterns of these proteins.
Clearly, integrated analysis by combining sequential, structural, interactomic analysis of the T3SS structural proteins revealed the abundance of protein intrinsic disorder in this system, identified specific patterns of protein-protein interaction, and discovered novel binding motifs in multiple T3SS structural proteins. The results are expected to facilitate further studies on the manipulation of inter-molecular interaction, disruption of structural integrity of T3SS, and selection of drug target.
Acknowledgment
BX is grateful to A. K. Dunker for the application of computation resources. The work was supported by the start-up funding from the Department of Cell Biology, Microbiology and Molecular Biology and College of Arts and Sciences at the University of South Florida to BX.
References
- 1. Hueck, C.J. Type III protein secretion systems in bacterial pathogens of animals and plants. (1998) Microbiol Mol Biol Rev 62(2): 379-433.
- 2. Ghosh, P. Process of protein transport by the type III secretion system. (2004) Microbiol Mol Biol Rev 68(4): 771-795.
- 3. Kuhle, V., Hensel, M. Cellular microbiology of intracellular Salmonella enterica: functions of the type III secretion system encoded by Salmonella pathogenicity island 2. (2004) Cell Mol Life Sci 61(22): 2812-2826.
- 4. Sun, G.W., Gan, Y.H. Unraveling type III secretion systems in the highly versatile Burkholderia pseudomallei. (2010) Trends Microbiol 18(12): 561-568.
- 5. Silva-Herzog, E., Detweiler, C.S. Salmonella enterica replication in hemophagocytic macrophages requires two type three secretion systems. (2010) Infect Immun 78(8): 3369-3377.
- 6. Diepold, A., Armitage, J.P. Type III secretion systems: the bacterial flagellum and the injectisome. (2015) Philos Trans R Soc Lond B Biol Sci 370(1679).
- 7. Perna, N.T., Plunkett, G., Burland, V., et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. (2001) Nature 409(6819): 529-533.
- 8. Hayashi, T., Makino, K., Ohnishi, M., et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. (2001) DNA Res 8(1): 11-22.
- 9. Kubori, T., Matsushima, Y., Nakamura, D., et al. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. (1998) Science 280(5363): 602-605.
- 10. Aizawa, S.I. Bacterial flagella and type III secretion systems. (2001) FEMS Microbiol Lett 202(2): 157-164.
- 11. Tamano, K., Aizawa, S., Katayama, E., et al. Supramolecular structure of the Shigella type III secretion machinery: the needle part is changeable in length and essential for delivery of effectors. (2000) Embo J 19(15): 3876-3887.
- 12. Kubori, T., Sukhan, A., Aizawa, S.I., et al. Molecular characterization and assembly of the needle complex of the Salmonella typhimurium type III protein secretion system. (2000) Proc Natl Acad Sci U S A 97(18): 10225-10230.
- 13. Kimbrough, T.G., Miller, S.I. Contribution of Salmonella typhimurium type III secretion components to needle complex formation. (2000) Proc Natl Acad Sci U S A 97(20): 11008-11013.
- 14. Demers, J.P., Sgourakis, N.G., Gupta, R., et al. The common structural architecture of Shigella flexneri and Salmonella typhimurium type three secretion needles. (2013) PLoS Pathog 9(3): e1003245.
- 15. Abrusci, P., Vergara-Irigaray, M., Johnson, S., et al. Architecture of the major component of the type III secretion system export apparatus. (2013) Nat Struct Mol Biol 20(1): 99-104.
- 16. Yu, X.J., McGourty, K., Liu, M., et al. pH sensing by intracellular Salmonella induces effector translocation. (2010) Science 328(5981): 1040-1043.
- 17. Deng, W., Li, Y., Hardwidge, P.R., et al. Regulation of type III secretion hierarchy of translocators and effectors in attaching and effacing bacterial pathogens. (2005) Infect Immun 73(4): 2135-2146.
- 18. Botteaux, A., Sory, M.P., Biskri, L., et al. MxiC is secreted by and controls the substrate specificity of the Shigella flexneri type III secretion apparatus. (2009) Mol Microbiol 71(2): 449-460.
- 19. Cherradi, Y., Schiavolin, L., Moussa, S., et al. Interplay between predicted inner-rod and gatekeeper in controlling substrate specificity of the type III secretion system. (2013) Mol Microbiol 87(6): 1183-1199.
- 20. Raymond, B., Young, J.C., Pallett, M., et al. Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. (2013) Trends Microbiol 21(8): 430-441.
- 21. Bliska, J.B., Wang, X., Viboud, G.I., et al. Modulation of innate immune responses by Yersinia type III secretion system translocators and effectors. (2013) Cell Microbiol 15(10): 1622-1631.
- 22. Deslandes, L., Genin, S. Opening the Ralstonia solanacearum type III effector tool box: insights into host cell subversion mechanisms. (2014) Curr Opin Plant Biol 20: 110-117.
- 23. Ashida, H., Mimuro, H., Sasakawa, C. Shigella manipulates host immune responses by delivering effector proteins with specific roles. (2015) Front Immunol 6:219.
- 24. Zychlinsky, A., Kenny, B., Menard, R., et al. IpaB mediates macrophage apoptosis induced by Shigella flexneri. (1994) Mol Microbiol 11(4): 619-627.
- 25. Hilbi, H., Moss, J.E., Hersh, D., et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. (1998) J Biol Chem 273(49): 32895-32900.
- 26. Boch, J., Bonas, U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. (2010) Annu Rev Phytopathol 48: 419-436.
- 27. O'Connell, C.B., Creasey, E.A., Knutton, S., et al. SepL, a protein required for enteropathogenic Escherichia coli type III translocation, interacts with secretion component SepD. (2004) Mol Microbiol 52(6): 1613-1625.
- 28. Martinez-Argudo, I., Blocker, A.J. The Shigella T3SS needle transmits a signal for MxiC release, which controls secretion of effectors. (2010) Mol Microbiol 78(6): 1365-1378.
- 29. Kresse, A.U., Beltrametti, F., Muller, A., et al. Characterization of SepL of enterohemorrhagic Escherichia coli. (2000) J Bacteriol 182(22): 6490-6498.
- 30. Kubori, T., Galan, J.E. Salmonella type III secretion-associated protein InvE controls translocation of effector proteins into host cells. (2002) J Bacteriol 184(17): 4699-4708.
- 31. Linares, J.F., Lopez, J.A., Camafeita, E., et al. Overexpression of the multidrug efflux pumps MexCD-OprJ and MexEF-OprN is associated with a reduction of type III secretion in Pseudomonas aeruginosa. (2005) J Bacteriol 187(4): 1384-1391.
- 32. Hachani, A., Biskri, L., Rossi, G., et al. IpgB1 and IpgB2, two homologous effectors secreted via the Mxi-Spa type III secretion apparatus, cooperate to mediate polarized cell invasion and inflammatory potential of Shigella flexenri. (2008) Microbes Infect 10(3): 260-268.
- 33. Namdari, F., Hurtado-Escobar, G.A., Abed, N., et al. Deciphering the roles of BamB and its interaction with BamA in outer membrane biogenesis, T3SS expression and virulence in Salmonella. (2012) PLoS One 7(11): e46050.
- 34. Roehrich, A.D., Guillossou, E., Blocker, A.J., et al. Shigella IpaD has a dual role: signal transduction from the type III secretion system needle tip and intracellular secretion regulation. (2013) Mol Microbiol 87(3): 690-706.
- 35. Liu, Z.Y., Zou, L.F., Xue, X.B., et al. HrcT is a key component of the type III secretion system in Xanthomonas spp. and also regulates the expression of the key hrp transcriptional activator HrpX. (2014) Appl Environ Microbiol 80(13): 3908-3919.
- 36. Gong, H., Vu, G.P., Bai, Y., et al. Differential expression of Salmonella type III secretion system factors InvJ, PrgJ, SipC, SipD, SopA and SopB in cultures and in mice. (2010) Microbiology 156(Pt 1):116-127.
- 37. Baron, C. Antivirulence drugs to target bacterial secretion systems. (2010) Curr Opin Microbiol 13(1): 100-105.
- 38. Keyser, P., Elofsson, M., Rosell, S., et al. Virulence blockers as alternatives to antibiotics: type III secretion inhibitors against Gram-negative bacteria. (2008) J Intern Med 264(1): 17-29.
- 39. Allen, R.C., Popat, R., Diggle, S.P., et al. Targeting virulence: can we make evolution-proof drugs? (2014) Nat Rev Microbiol 12(4): 300-308.
- 40. Clatworthy, A.E., Pierson, E., Hung, D.T. Targeting virulence: a new paradigm for antimicrobial therapy. (2007) Nat Chem Biol 3(9): 541-548.
- 41. Coates, A.R., Halls, G., Hu, Y. Novel classes of antibiotics or more of the same? (2011) Br J Pharmacol 163(1): 184-194.
- 42. Marra, A. Targeting virulence for antibacterial chemotherapy: identifying and characterising virulence factors for lead discovery. (2006) Drugs R D 7(1): 1-16.
- 43. McShan, A.C., De Guzman, R.N. The bacterial type III secretion system as a target for developing new antibiotics. (2015) Chem Biol Drug Des 85(1): 30-42.
- 44. Gu, L., Zhou, S., Zhu, L., et al. Small-Molecule Inhibitors of the Type III Secretion System. (2015) Molecules 20(9): 17659-17674.
- 45. Tosi, T., Pflug, A., Discola, K.F., et al. Structural basis of eukaryotic cell targeting by type III secretion system (T3SS) effectors. (2013) Res Microbiol 164(6): 605-619.
- 46. Burkinshaw, B.J., Strynadka, N.C. Assembly and structure of the T3SS. (2014) Biochim Biophys Acta 1843(8): 1649-1663.
- 47. Chatterjee, S., Zhong, D., Nordhues, B.A., et al. The crystal structures of the Salmonella type III secretion system tip protein SipD in complex with deoxycholate and chenodeoxycholate. (2011) Protein Sci 20(1): 75-86.
- 48. Dasanayake, D., Richaud, M., Cyr, N., et al. The N-terminal amphipathic region of the Escherichia coli type III secretion system protein EspD is required for membrane insertion and function. (2011) Mol Microbiol 81(3): 734-750.
- 49. Chaudhury, S., Battaile, K.P., Lovell, S., et al. Structure of the Yersinia pestis tip protein LcrV refined to 1.65 A resolution. (2013) Acta Crystallogr Sect F Struct Biol Cryst Commun 69(Pt 5): 477-481.
- 50. Banerjee, A., Dey, S., Chakraborty, A., et al. Binding mode analysis of a major T3SS translocator protein PopB with its chaperone PcrH from Pseudomonas aeruginosa. (2014) Proteins 82(12): 3273-3285.
- 51. Gazi, A.D., Bastaki, M., Charova, S.N., et al. Evidence for a coiled-coil interaction mode of disordered proteins from bacterial type III secretion systems. (2008) J Biol Chem 283(49): 34062-34068.
- 52. Hu, W., Anand, G., Sivaraman, J., et al. A disordered region in the EvpP protein from the type VI secretion system of Edwardsiella tarda is essential for EvpC binding. (2014) PLoS One 9(11):e110810.
- 53. Xue, B., Dunker, A.K., Uversky, V.N. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. (2012) J Biomol Struct Dyn 30(2): 137-149.
- 54. Xue, B., Williams, R.W., Oldfield, C.J., et al. Viral disorder or disordered viruses: do viral proteins possess unique features? (2010) Protein Pept Lett 17(8): 932-951.
- 55. Xue, B., Mizianty, M.J., Kurgan, L., et al. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. (2012) Cell Mol Life Sci 69(8): 1211-1259.
- 56. Xue, B., Ganti, K., Rabionet, A., et al. Disordered interactome of human papillomavirus. (2014) Curr Pharm Des 20(8): 1274-1292.
- 57. Fan, X., Xue, B., Dolan, P.T., et al. The intrinsic disorder status of the human hepatitis C virus proteome. (2014) Mol Biosyst 10(6): 1345-1363.
- 58. Xue, B., Blocquel, D., Habchi, J., et al. Structural disorder in viral proteins. (2014) Chem Rev 114(13): 6880-6911.
- 59. Butler, C.L., Lucas, O., Wuchty, S., et al. Identifying novel cell cycle proteins in Apicomplexa parasites through co-expression decision analysis. (2014) PLoS One 9(5): e97625.
- 60. Dolan, P.T., Roth, A.P., Xue, B., et al. Intrinsic disorder mediates hepatitis C virus core-host cell protein interactions. (2015) Protein Sci 24(2): 221-235.
- 61. Rosqvist, R., Hakansson, S., Forsberg, A., et al. Functional conservation of the secretion and translocation machinery for virulence proteins of yersiniae, salmonellae and shigellae. (1995) Embo J 14(17):4187-4195.
- 62. Hermant, D., Menard, R., Arricau, N., et al. Functional conservation of the Salmonella and Shigella effectors of entry into epithelial cells. (1995) Mol Microbiol 17(4): 781-789.
- 63. Chatterjee, S., Chaudhury, S., McShan, A.C., et al. Structure and biophysics of type III secretion in bacteria. (2013) Biochemistry 52(15): 2508-2517.
- 64. Szklarczyk, D., Franceschini, A., Wyder, S., et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. (2015) Nucleic Acids Res 43: 447-452.
- 65. Salwinski, L., Miller, C.S., Smith, A.J., et al. The Database of Interacting Proteins: 2004 update. (2004) Nucleic Acids Res 32: D449-451.
- 66. Romero, P., Obradovic, Z., Li, X., et al. Sequence complexity of disordered protein. (2001) Proteins 42(1): 38-48.
- 67. Xue, B., Dunbrack, R.L., Williams, R.W., et al. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. (2010) Biochim Biophys Acta 1804(4): 996-1010.
- 68. Li, J., Feng, Y., Wang, X., et al. An Overview of Predictors for Intrinsically Disordered Proteins over 2010-2014. (2015) Int J Mol Sci 16(10): 23446-23462.
- 69. Xue, B., Oldfield, C.J., Van, Y.Y., et al. Protein intrinsic disorder and induced pluripotent stem cells. (2012) Mol Biosyst 8(1): 134-150.
- 70. Xu, K., Uversky, V.N., Xue, B. Local flexibility facilitates oxidization of buried methionine residues. (2012) Protein Pept Lett 19(6): 688-697.
- 71. Xue, B., Brown, C.J., Dunker, A.K., et al. Intrinsically disordered regions of p53 family are highly diversified in evolution. (2013) Biochim Biophys Acta 1834(4): 725-738.
- 72. Malaney, P., Pathak, R.R., Xue, B., et al. Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. (2013) Sci Rep 3: 2035.
- 73. Xue, B., Uversky, V.N. Intrinsic disorder in proteins involved in the innate antiviral immunity: another flexible side of a molecular arms race. (2014) J Mol Biol 426(6): 1322-1350.
- 74. Na, I., Reddy, K.D., Breydo, L., et al. A putative role of the Sup35p C-terminal domain in the cytoskeleton organization during yeast mitosis. (2014) Mol Biosyst 10(4): 925-940.
- 75. Brunquell, J., Yuan, J., Erwin, A., et al. DBC1/CCAR2 and CCAR1 Are Largely Disordered Proteins that Have Evolved from One Common Ancestor. (2014) Biomed Res Int 2014: 418458.
- 76. Yuan, J., Xue, B. Role of structural flexibility in the evolution of emerin. (2015) J Theor Biol 385: 102-111.
- 77. Petersen, B., Petersen, T.N., Andersen, P., et al. A generic method for assignment of reliability scores applied to solvent accessibility predictions. (2009) BMC Struct Biol 9: 51.
- 78. Finn, R.D., Coggill, P., Eberhardt, R.Y., et al. The Pfam protein families database: towards a more sustainable future. (2016) Nucleic Acids Res 44(D1): D279-285.
- 79. Oldfield, C.J., Cheng, Y., Cortese, M.S., et al. Coupled folding and binding with alpha-helix-forming molecular recognition elements. (2005) Biochemistry 44(37): 12454-12470.
- 80. Cheng, Y., Oldfield, C.J., Meng, J., et al. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. (2007) Biochemistry 46(47): 13468-13477.
- 81. Dosztanyi, Z., Meszaros, B., Simon, I. ANCHOR: web server for predicting protein binding regions in disordered proteins. (2009) Bioinformatics 25(20): 2745-2746.










