
Formulation and Evaluation of Self-Nanoemulsifying Powder of Ezetimibe
Suresh K. Dumaniya, Chhaganbhai N. Patel
Affiliation
Shri Sarvajanik Pharmacy College, Near Arvind Baug, Mehsana-384001, Gujarat, India.
Corresponding Author
Shailesh T. Prajapati, Department of Pharmaceutics, Shri Sarvajanik Pharmacy College, Near Arvind Baug, Mehsana-384001, Gujarat, India, E-mail: stprajapati@gmail.com
Citation
Shailesh TP. et al. Formulation and Evaluation of Self-Nanoemulsifying Powder of Ezetimibe. (2014) J Pharma Sci Drug Des 1(1): 4-11
Copy rights
©2014 Shailesh TP. This is an Open access article distributed under the terms of Creative Commons Attribution 4.0 International License.
Keywords
Ezetimibe; Self nanoemulsifying powder; Pseudo ternary phase diagram; Solubility study
Abstract
Ezetimibe (EZT) is the selective cholesterol absorption inhibitor. It is indicated for the treatment of primary hypercholesterolemia. The current investigation was aimed to formulation and evaluation of self nanoemulsifying powder (SNEP) for EZT to enhance solubility and dissolution rate and this will leads to minimize the variability in absorption. Solubility of drug was determined in different vehicles. Pseudo ternary phase diagram generated using Acrysol EL 135, Tween 80 and Trancutol P. The SNEP was prepared by adsorbing the optimized liquid SNEDDS on to Fujicalin as carrier. The SNEP characterized by micromeritic properties, differential scanning studies, percentage transmittance, emulsification time and zeta potential. The optimized formulation had shown the smallest particle size, less emulsification time, good %transmittance and good in vitro release. The in vitro dissolution rate and ex vivo permeation rate of the drug from the SNEP was significantly higher than that of the plain drug and its suspension respectively.
Introduction
Formulation of poorly aqueous soluble drug is a challenging job to the pharmaceutical scientists as result of modern drug discovery technique and oral delivery of such drugs is frequently associated with low bioavailability; high inter subject variability and lack of dose proportionality. The formulation technique plays an important role in overcoming this shortcoming of this poorly water soluble drugs, various formulation strategies are including use of surfactants, pulverization, crystal polymorphism selection, salt formation, solid dispersion, complex formation agent like cyclodextrin, emulsion, microemulsion, liposome, nanoparticles, micro and nanospheres, lipid carriers, use of prodrug, drug derivatization, solution phase studies and permeation enhancers to improve the dissolution rate of the drug[1,2].
EZT is a selective cholesterol absorption inhibitor that effectively blocks intestinal absorption of dietary and biliary cholesterol and used to treat patients with hypercholesterolemia. Logarithm of partition coefficient [log P (octanol/water)] value of EZT (Figure 1) is 4.5 and it comes under BCS class II drug. Poor solubility of the drug is associated with poor dissolution rate and thus low oral bioavailability[3]. EZT pharmacokinetics exhibited moderate inter-subject variability, with coefficients of variation (CVs) ranging from 34% to 43% and 32% to 37% for Cmax and AUC, respectively. The absolute bioavailability of EZT (Figure 1a) cannot be determined, as the compound is virtually insoluble in aqueous media suitable for injection[4].
SNEDDS is an isotropic mixture composed of oil, surfactant, co-surfactant and drug. It can readily disperse in the aqueous environment of the gastrointestinal tract to form a fine oil-in-water emulsion with a droplet size less than 100nm under gentle agitation for improving the oral bioavailability of poorly water-soluble drugs[5,6]. Liquid SNEDDS (Figure 1b) are filled into soft gels or sealable hard gelatin capsules which have some shortcomings like these dosage forms may be inconvenient to use and incompatibility problems with the capsule shells are usual. Incorporation of a liquid self-emulsifying formulation into a solid dosage form may combine the advantages of SNEDDS with those of a solid dosage form and overcome the disadvantages of liquid formulations described above[7].Dixit R et.al.[8] prepared self nano-emulsifying granules of ezetimibe using various modified oils, surfactant and co-surfactant mixtures. The self nanoemulsifying systems were formulated into free flowing self nanoemulsifying granules (SNG) using varying proportions of hydrophilic colloidal silicon dioxide as an adsorbing agent. The SNG filled into hard gelatine capsules showed two to threefold increase in the dissolution rate as compared to plain drug filled capsules signifying its potential in improved delivery of lipophilic drugs[8]. Many research works are reported on SNEDDS of Ezetimibe but not much work reported on solid SNEDDS of Ezetimibe. Hence, aim of present investigation was to develop SNEDDS with Acrysol EL 135 as oil, Tween 80 as surfactant and Transcutol P as co-surfactant and convert it in to a powder by using various adsorbent like Fujicalin, Silicon dioxide etc.
Materials and Methods
Materials
EZT was a generous gift from Astron Research Lab, Ahmedabad, India. Transcutol P (Diethylene glycol monoethyl ether), Plurol Oleique CC 497 (Polyglyceryl-3 dioleate), Lauroglycol (Propylene glycol monolaurate) were kindly provided as a gift sample by Gattefosse, India. Capmul MCM (Glycerol mono-dicaprylate), Accono C-80 (Polyoxyethylene 80 coconut glycerides) were received as a gift sample from Abitec Corporation, US. Acrysol EL 135 (Polyoxyl 35 castor oil), Acrysol K 150 (Polyoxyl 50-hydrogenated castor oil), Acrysol K 140 (Poyoxyl 40-hydrogenated castor oil) were gifted from Corel Pharma Chem, Ahmedabad, India. Fujicalin (Dibasic calcium phosphate) was generously donated by Fuji Chemicals, Tokyo, Japan. Tween 80, Tween 20, Propylene glycol were purchased from Finar Chemical Limited, Ahmedabad, India. Polyethylene glycol 200 (PEG 200), Polyethylene glycol 400 (PEG 400) were procured from S. D. Fine Chemical Limited, Mumbai, India. All other reagents and solvents used were of analytical grade.
Selection of self nanoemulsified drug delivery system components
Solubility studies:Solubility of EZT was determined in various modified oils, surfactants and cosurfactants. Two mL of each component was taken in screw cap vials with known quantity of excess drug. A vortex mixture (Spinix, India) was used to facilitate the solubilisation. Sealed vials were kept on isothermal mechanical shaker at 40 ± 2°C for 72 hours. After equilibrium, each test tube was centrifuged at 6000 rpm for 20 minute using a centrifuge (R-8C, Remi, India). Supernatant was filtered through membrane filter using 0.45μm filter disk[8]. Filtered solution was appropriately diluted with methanol, and UV absorbance was measured at 233 nm. Concentration of dissolved drug was determined using standard equation.
Surfactant (Emulsification study): Different surfactants were screened for its emulsification ability selected in oil phase. Surfactant selection was done on the basis of % transparency and ease of emulsification. Briefly, 300 mg of surfactant was added to 300 mg of oil phase. The mixture was heated at 50°C for the homogenization of the components. Each mixture, 50 mg, was then diluted with 50 mL distilled water in glass stopper conical flask. Ease of emulsification was judged by the number of flask inversions required to yield homogenous emulsion were allowed to stand for 2 h and their %transparency was evaluated at 650 nm by a double-beam UV spectrophotometer using distilled water as a blank[9]. Emulsions were furthermore observed visually for any turbidity or phase separation.
Co-surfactant (Emulsification study): The screening of the co-surfactant was conducted on the basis of %transparency and ease of emulsification. Mixtures of 100 mg of the co-surfactant, 200 mg of the selected surfactant, and 300 mg of the selected oil were prepared and evaluated in a similar fashion as described in the above section on surfactants[9].
Drug excipients compatibility studies (FTIR studies): FTIR spectroscopy helps to determine any chemical interaction between drug and excipients used in formulation. The FTIR spectra for EZT and self nanoemulsifying powder mixture preparations were obtained using FTIR-8400S spectrophotometer (Shimadzu, Japan) in the range of 4000-400 cm-1.
Construction of the pseudo ternary phase diagrams
On the basis of solubility and emulsification study Acrysol EL 135, Tween 80 and Transcutol P were selected as oil, surfactant and co-surfactant respectively. To determine the concentration of components for the existing range of the SNEDDS, a pseudo ternary phase diagram was constructed using a water titration method at ambient temperature (25°C). The Surfactant and co-surfactant were mixed (Smix) in different volume ratios (1:1, 1:2, 1:3, 2:1, 3:1). For each phase diagram, oil and specific Smix ratio was mixed well in different volume ratios ranging from 1:9 to 9:1. Sixteen different combinations of oil and Smix (1:9, 1:8, 1:7, 1:6, 1:5, 1:3.5, 1:3, 3:7, 1:2, 2:8, 4:6, 5:5, 6:4, 7:3, 8:2 and 9:1) were titrated with water by dropwise addition under gentle agitation[10,2]. Physical state of the nanoemulsion was marked on a pseudo three component phase diagram with one axis representing the aqueous phase, one representing oil and the third representing a mixture of surfactant and co-surfactant at fixed volume ratio (Smix) using Sigma Plot Software (Version 12.0). To determine the effect of drug addition on the nanoemulsion boundary, phase diagrams were also constructed in the presence of drug using drug-enriched oil as the oil phase.
Preparation of the liquid self nanoemulsified formulations
EZT (10 mg) was added in accurately weighed amount of oil into a screw capped glass vial and heated in a water bath at 37°C. The surfactant and co-surfactant were added to the oil mixture using positive displacement pipette and stirred with magnetic bar. The formulation were further sonicated (Ultrasonic cleaner EN-30-US, Electroquip, India) for 15 min and stored at room temperature until their use in subsequent studies. The compositions for different batch LS1 to LS4 are shown in Table 1.
Table 1: Solubility studies of Ezetimibe in various vehicles
| Sr. No. | Vehicles | Role of Excipients | Avg. Solubility (mg/mL) ± SD (n=3) |
|---|---|---|---|
| 1 | Capmul MCM | Oil | 032.54 ± 0.494 |
| 2 | Transcutol P | Co-Surfactant | 219.90 ± 0.940 |
| 3 | Plurol Oleique CC 497 | Oil | 004.83 ± 0.473 |
| 4 | Lauroglycol | Surfactant | 004.03 ± 0.091 |
| 5 | Acconon C-80 | Co-Surfactant | 041.13 ± 0.083 |
| 6 | Tween 80 | Surfactant | 051.39 ± 0.742 |
| 7 | Tween 20 | Surfactant | 028.86 ± 0.431 |
| 8 | Polyethylene glycol 200 | Co-Surfactant | 2.65 ± 0.077 |
| 9 | Propylene glycol | Co-Surfactant | 1.55 ± 0.220 |
| 10 | Polyethylene glycol 400 | Co-Surfactant | 9.21 ± 0.296 |
| 11 | Acrysol EL 135 | Oil | 132.38 ± 2.404 |
| 12 | Acrysol K 150 | Oil | 088.28 ± 1.647 |
| 13 | Acrysol K 140 | Oil | 045.68 ± 0.035 |
*(Mean ± SD; n = 3)
Evaluation of liquid SNEDDS formulations
Emulsification time: The emulsification time (the time for a preconcentrate to form a homogeneous mixture upon dilution) was monitored by visually observing the disappearance of SNEDDS and the final appearance of the nanoemulsion in triplicate. A USP dissolution apparatus II (TDT-08L, Electrolab, Mumbai, India) was employed with 500 mL water, and with a paddle speed of 50 rpm at 37°C. The SNEDDS (1mL) was added drop wise to the medium by a dropping pipette and the time required for the disappearance of the SNEDDS was recorded[11].
Globule size analysis and zeta potential: Formulations (LS1 to LS4) each of 1 ml were diluted with 100 ml of distilled water in a volumetric flask. The volumetric flask was inverted twice to ensure complete dispersion of the formulation. After ensuring complete dispersion of the formulation the globule size and zeta potential of resultant microemulsion was determined by photon correlation spectroscopy using a (Zetasizer, Malvern Instruments, USA). Light scattering was monitored at 25°C at 90° angle.
In-vitro drug release study: The in vitro drug release of EZT from the LS1 to LS4 SNEDDS was performed using USP dissolution Apparatus II (TDT-08L, Electrolab, Mumbai, India). Hard gelatin capsules, size "00" filled with preconcentrate (equivalent to 10 mg EZT) were put into 500ml of 0.45% SLS 0.05 M acetate buffer pH 4.5, at 37 ± 0.5°C with a 50 rpm rotating speed. Samples (10ml) were withdrawn at regular time intervals (5, 10, 15, 30, 45, and 60 min) and filtered using a whatman filter paper. An equal volume of the dissolution medium was added to maintain the volume constant. The drug content of the samples was assayed using UV visible spectrophotometric method. All measurements were done in triplicate.
Determination of drug content:EZT from SNEDDS formulation was extracted in methanol using sonication technique. The solutions were filtered, using whatman filter paper. The methanolic extract was analyzed for the EZT content spectrophotometrically (UV-1800, Shimadzu, Japan) at 233 nm using standard curve.
Percentage transmittance: Percentage transmittance of the prepared nanoemulsion formulations was determined spectrophotometrically using UV-visible spectrophotometer (Shimadzu, Japan). One ml of the formulation was diluted 100 times using methanol and analyzed at 233 nm using methanol as blank[12].
Preparation of self nanoemulsifying powder
SNEP was prepared by mixing optimized LS1 liquid SNEDDS containing EZT with Fujicalin adsorbent carrier. In brief Fujicalin® was added over liquid SNEDDS aliquot (equivalent to 10 mg EZT) contained in broad porcelain dish. After each addition, mixture was homogenized using a glass rod until a uniform distribution, free flowing powder was obtained of formulation[13]. Resultant damp mass was passed through 60# sieve and dried at ambient temperature. Then, the powder was filled in a "00" size hard gelatin capsule shell and stored for further use. Evaluation of self nanoemulsifying powder
Flow properties of self nanoemulsifying powder: Angle of repose (θ) of SNEP was determined by funnel method. Bulk density, Compressibility Index and Hausner ratio were also determined[14].
Emulsification time, Globule size analysis and zeta potential, Determination of drug content and % Transmittance: SNEP were also characterized emulsification time, globule size analysis and zeta potential, determination of drug content and %transmittance as described for liquid SMEDDS.
Solid state characterization of Self nanoemulsifying powder: The physical state of EZT in SNEP was characterized by the DSC (DSC-60, Shimadzu Corporation, Tokyo, Japan). Thermograms of pure drug and SNEP were obtained using DSC. The samples (about 3 mg) were placed in standard aluminium pans and heated at constant rate 10ºC/min, over a temperature range of 30ºC to 200ºC. Inert atmosphere was maintained by purging nitrogen at the flow rate of 20 mL/min.
In-vitro drug release study: The in vitro drug release of EZT from the SNEP was performed using USP dissolution apparatus II (TDT-08L, Electrolab, Mumbai, India). Hard gelatin capsules, size "00" filled with SNEP (equivalent to 10 mg EZT) and pure drug (10 mg) separately were put into each 500 ml 0.45% SLS 0.05 M acetate buffer pH 4.5, at 37 ± 0.5°C with a 50 rpm rotating speed. Samples (10ml) were withdrawn at regular time intervals (5, 10, 15, 30, 45, and 60 min) and filtered using a 0.45 μm filter. An equal volume of the dissolution medium was added to maintain the volume constant. The drug content of the samples was assayed using UV visible spectrophotometric method. All measurements were done in triplicate.
Ex vivo intestinal permeability study: All experiments and protocols described in this study were approved by the Institutional Animal Ethics Committee (IAEC) and all experiments were conducted as per the norms of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Social Justice and Empowerment, Government of India. Male Wister rats (250-300g) were sacrificed by CO2 inhalation method. Intestine was isolated and cleaned properly. One millilitre of EZT reconstituted solution of SNEP and plain drug suspension sample (5mg/ml) was filled into the intestine individually which was tied at both the end. The tissue was placed in an organ bath with continuous aeration at 37°C. The receptor compartment (organ tube) was filled with Krebs ringer solution (KRB)[14]. At predetermined time intervals, samples were withdrawn from the receptor compartment. Fresh buffer was used to replenish the receptor compartment. The samples were analysed spectrophotometrically for the content of EZT. The percent diffusion was calculated and plotted against time. The study was also repeated with EZT plain suspension and the results were compared. All the experiments were done in triplicate.
Short term stability study
The optimized formulation was subjected to short term stability studies as per ICH guidelines conditions Q1C in order to assess its chemical and physical stability under the following conditions: 40ºC ± 2ºC and 75% ± 5% relative humidity in stability chamber (Thermolab, Bombay). The stability studies were followed for 1 month. The stored samples were evaluated for appearance, colour, EZT content, self emulsification time and the dissolution profile of the EZTS-SMEDDS hard gelatine capsules.
Results and Discussion
Solubility study (screening of oil)
The oil represents one of the most essential excipients in the SNEDDS formulation. Identifying the suitable oil can solubilize the necessary dose of the lipophilic drug and it can improve the fraction of lipophilic drug transported via the intestinal lymphatic system, thereby increasing absorption from the GI tract[15]. The solubility of EZT in various oils and surfactants solution is presented in Table 2. Among the various oils that were screened, Acrysol EL 135 could solubilize the largest amount of EZT (132.38 ± 2.404 mg/ml). The selection of the surfactant or co-surfactant in the further study was governed by the emulsification efficiency rather than the ability to solubilize EZT.
Table 2: Emulsification efficiency of various surfactants and co-surfactants
| Surfactants/Co-surfactants | % Transparency | No. of inversions |
|---|---|---|
| Tween 80 | 98.92 ± 0.035 | 5 |
| Tween 20 | 98.10 ± 0.141 | 15 |
| Transcutol P | 99.84 ± 0.070 | 5 |
| Plurol Oleique | 88.55 ± 0.070 | 25 |
| PEG 200 | 98.11 ± 0.014 | 35 |
| PEG 400 | 99.57 ± 0.212 | 30 |
| Capmul MCM C-8 | 80.15 ± 0.212 | 40 |
*(Mean ± SD; n = 3)
Preliminary screening of surfactants
Non-ionic surfactants are identified to be less toxic compared to ionic surfactants. They are usually accepted for oral ingestion[16]. Acrysol EL 135 exhibited maximum emulsification efficiency and maximum transmittance (98.92 ± 0.035%) with Tween 80 surfactants. While with Tween 20 showed poor emulsification property requiring higher number of flask inversions [Table 3]. The afore mentioned results suggested the use of Acrysol EL 135 as an oily phase with Tween® 80 as a surfactant for further study.
Table 3: Formulations at a different % v/v of oil, surfactant and co-surfactant
| Formulation code | Composition (% vol. /vol.) | ||
|---|---|---|---|
| Acrysol El 135 | Tween 80 | Transcutol P | |
| LS1 | 30 | 35 | 35 |
| LS2 | 34 | 33 | 33 |
| LS3 | 36 | 32 | 32 |
| LS4 | 40 | 30 | 30 |
Preliminary screening of co-surfactants
Addition of a co-surfactant to the surfactant containing formulation was reported to improve dispersibility and drug absorption from the formulation[17,18]. As depicted in Table 3, Acrysol EL 135 exhibited good emulsification with all co-surfactants, with Transcutol P showing the maximum transmittance (99.84 ± 0.070%) followed by PG (99.57 ± 0.212%). Herein, the solubility of the drug in different co-surfactants may judge the final selection. Results of the solubility study demonstrated in Table 2 inferred a higher solubility in Transcutol P. It is worthy to note that all dispersions exhibited an instantaneous emulsion formation with only five flask inversions [Table 3]. This could contend the importance of co-surfactant addition to the surfactant containing dispersions.
Drug excipients compatibility studies (FTIR studies)
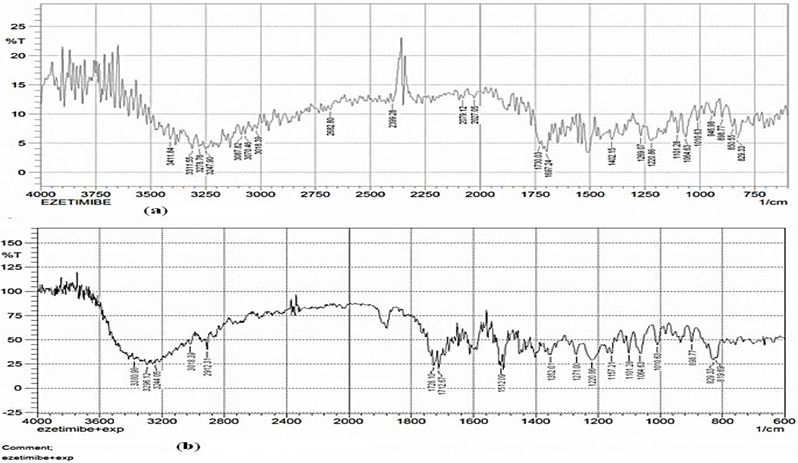
The FTIR spectra of EZT showed at 3411.84-3247.90 cm-1 (O-H, Alcohol/ Phenol str), 3087.82-3018.39 cm-1 (C-H, Aromatic str), 1697.24 cm-1 (N-C=O Amide str), and 1101.28 cm-1 (C-F bending). These peaks can be considered as characteristic peaks of Ezetimibe and were not affected and prominently observed in IR spectra of ezetimibe along with oil, surfactant co-surfactant and adsorbent Fujicalin. It was concluded that drug and excipient are compatible.
Pseudo ternary phase diagrams
To determine the spontaneity of SNEDDS to form the nanoemulsion within the GI conditions, the construction of pseudo ternary phase diagram considered to be important exercise. Ternary phase behaviour investigations help to choose the proper concentration of excipients i.e., oil proportion and optimum Smix ratio in the formulation to produce emulsions with good stability[19].
Due to low toxicity and less affected by pH and ionic strength changes of non-ionic or zwitterionic surfactants; they are frequently used for nanoemulsion formulation[5]. Thus for the present study, Tween 80 having hydrophilic–lipophilic balance (HLB) value of 15 and Transcutol P ctant and is also known to increase the permeability of drugs. The phase diagrams were constructed at surface having HLB of 4.2 was used as the surfactant and co-surfactant respectively. Transcutol P is a non-ionic co-surfatant/co T surfactant ratio of 1:1, 1:2, 1:3, 2:1 and 3:1(v/v). Phase diagram of different surfactant and co-surfactant ratio is shown in Figure 2. In present study Acrysol EL 135 as oil, Tween 80 as surfactant, and Transcutol P as co-surfactant showed broad region of nanoemulsion and hence quite promising. When co-surfactant was used along with the surfactant in 1:1 ratio [Figure 2(a)], a tremendous increase in the nanoemulsion region was observed along with the maximum amount of oil that could be emulsified and 63% (v/v) of oil could be emulsified using 27% (v/v) of Smix. When the concentration of surfactant Tween 80 was doubled or tripled, Smix ratio 1:2 or1:3 the gel like region was found to become large while the nanoemulsion region was decreased [Figure 2(b,c)]. While when the concentration of co-surfactant, was increased in the Smix from 1:1 to 1:2 [Figure 2(d)], slight change in the nanoemulsion region was observed. On further increasing the concentration of co-surfactant in the Smix from 1:2 to 1:3 [Figure 2(e)], it was observed that the nanoemulsion region remained almost same.
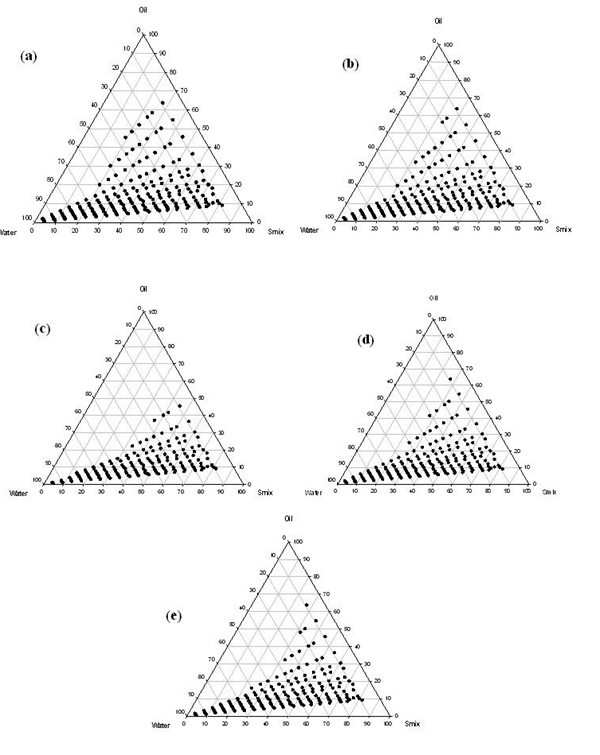
Figure 2: Acrysol EL 135 as oil, Tween 80 as surfactant, Transcutol P as co-surfactant. Ratio (% v/v) of surfactant to co-surfactant in a) 1:1, (b) 2:1, (c) 3:1, (d) 1:2, (e) 1:3 is shown. Dotted area shows oil/water nanoemulsion region
Co-surfactants are beneficial to form a nanoemulsion at a proper concentration range. However, an excessive amount of the co-surfactant will cause the system to become less stable for its intrinsic high aqueous solubility and lead to the droplet size increasing as a result of the expanding interfacial film[11]. Hence, the optimal ratio of surfactant to co-surfactant was selected to be 1:1.
Based on above results, a three component SNEDDS formulation was established containing 30% Acrysol EL 135 as oil (on the basis of the solubility study and required target amount of EZT, 10 mg), 35% Tween® 80 as the surfactant, and 35% Transcutol P as the co-surfactant (on the basis of phase diagrams). Four SNEDDS formulations were prepared and they evaluation parameters to be compared.
Evaluation of liquid SNEDDS formulations
Emulsification time: In SNEDDS, the primary means of self emulsification assessment is visual estimation. The efficiency of self emulsification could be estimated primarily by determining the rate of emulsification which is an important index for the assessment of the efficiency of emulsification that is the SNEDDS should disperse completely and quickly when subjected to aqueous dilution under mild agitation. The emulsification time study showed that the all four formulation employed could emulsify within range of 18 to 30 sec [Table 4].
Table 4: Emulsification time, Globule size, PDI, zeta potential and % Transmittance of liquid SNEDDS formulations
| Formulation code | Emulsification time* | Globule size in water* | Polydispersibility Index* | Zeta potential* | Percenrage transmittance* |
|---|---|---|---|---|---|
| LS1 | 18.6 ± 0.5 | 011.10 ±1.76 | 0.177 ±0.006 | -24.65 ± 0.05 | 97.490 ± 0.002 |
| LS2 | 22.0 ± 1.0 | 022.28 ±0.64 | 0.162±0.010 | -17.56 ± 0.04 | 93.972 ± 0.001 |
| LS3 | 26.0 ± 1.0 | 034.58 ±0.89 | 0.230±0.007 | -09.74 ± 0.01 | 88.920 ± 0.001 |
| LS4 | 30.0 ± 1.0 | 046.41 ±0.77 | 0.253±0.008 | -04.72 ± 0.02 | 81.470 ± 0.001 |
*(Mean ± SD; n = 3)
Globule size analysis and zeta potential: The globule size of the emulsion is a crucial factor in self emulsification performance because it determines the rate and extent of drug release as well as absorption[20]. Formulation LS1 was found to have the smallest globule size and least polydispersity index (PDI) of 11.10 ± 1.76 nm (mean ± SD, n = 3) and 0.117 respectively [Table 4]. Since PDI of the developed nanoemulsion formulations in the present study was small, this indicates uniformity in the size distribution of the dispersed oil globules.
Zeta potential signifies degree of repulsion between neighbouring, like charged particles in dispersion; it can be related to the stability of colloidal dispersions. The zeta potential of the optimized formulation was -24.65 ± 0.05 (mean ± SD, n = 3). In general, the zeta potential value of ± 30 mV is sufficient for the stability of a nanoemulsion. Negative values of zeta potential of the optimized formulations showed that the formulations were negatively charged and high values of zeta potential of all the formulations denoted stability of the system. The high negative charge of the prepared nanoemulsion is probably due to the anionic groups of the fatty acids and glycols present in the oil, surfactant and co-surfactant.
In-vitro drug release study: Dissolution studies were performed for the liquid SNEDDS formulations in 0.45% SLS 0.05M acetate buffer pH 4.5. There is no any significant difference in dissolution of four SNEDDS formulation. The all SNEDDS formulation was found to release about 100% of the drug at the end of 15 min [Figure 3].
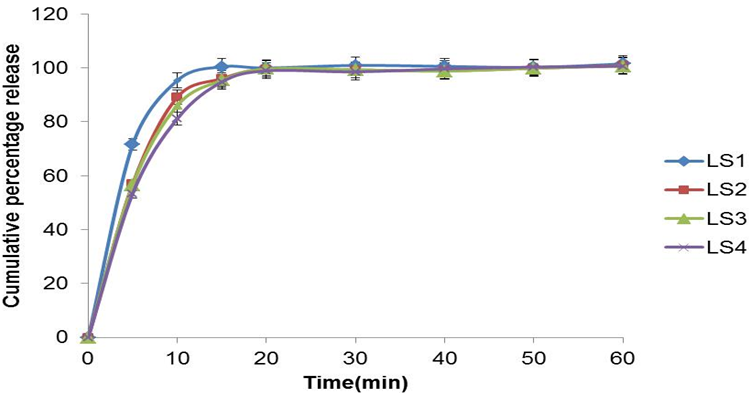
This could be attributed to the fact that the quantitative release of the EZT from the SNEDDS is droplet size dependent. This suggests that larger interfacial area present in emulsions with smaller drops, promotes rapid drug release. Therefore drug release rate of the drug was increased by decreasing the nanoemulsions droplet size, suggesting that release rate of poorly water soluble drug like EZT could be controlled by judiciously selecting the mean droplet size in the carrier emulsion generated from SNEDDS.
% Transmittance: Formulation LS1 was found to have the highest percentage transmittance. A value of percentage transmittance closer to 100% signified that all of the optimized formulations were clear and transparent [Table 4]. Besides signifying clarity of the formulation, a percentage transmittance closer to 100% also implies that the size of the globules in the nanoemulsion formulation approximates the nanometer range.
Drug content:Irrespective of difference in composition the drug content of formulations LS1 to LS4 was found in range of 99.04 – 99.92%.
Evaluation of self nanoemulsifying powder
Flow properties of SNEP: SNEP was prepared to overcome the disadvantage associated with liquid SNEDDS. Hence to overcome the problem of stability and improve patient compliance formulation LS1 was adsorbed onto Fujicalin solid adsorbent carrier. Various micromeritic properties of SNEP of EZT are shown in [Table 5]. Results showed that SNEP has good flow properties.
Table 5: Flow properties of SNEP formulation
| Formulation Code | Flow Properties | Results * | Inferences |
|---|---|---|---|
| SS1 | Angle of Repose (degree) | 29.59 ± 0.625 | Good |
| Carr's Index (%) | 15.00 ± 0.915 | Good | |
| Hausner Ratio | 01.17 ± 0.026 | Good |
*(Mean ± SD; n = 3)
Globule size analysis and zeta potential and drug content: The z-average diameter and polydispersibility index of SNEP and liquid SNEDDS was shown in [Table 6]. As shown in table there were no more change in globule size, PDI and zeta potential of the liquid and solid formulation. Drug content was found within range 99.52 ± 0.083%.
Table 6: Emulsificationtime, Globulesize, PDI, zeta potential and % Transmittance of SNEP
| Formulation code | Emulsification time* | Globule size in water* | Polydispersibility Index* | Zeta potential* | Percentage transmittance* |
|---|---|---|---|---|---|
| SNEP | 26.6 ± 0.4 | 41.60 ±1.47 | 0.243 ± 0.008 | -23.85 ±0.31 | 94.256 ± 0.856 |
| LS1 | 18.6 ± 0.5 | 011.10 ±1.76 | 0.177 ± 0.006 | -24.65 ± 0.05 | 97.490 ± 0.002 |
*(Mean ± SD; n = 3)
% Transmittance: Percent transmittance of reconstituted SNEP was found to be 94.256 ± 0.856 [Table 6]. It indicates that reconstituted SNEP was found to be clear.
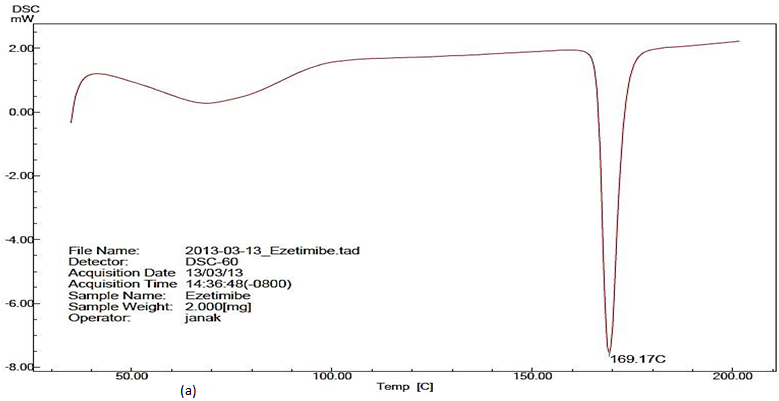
Differential scanning calorimeter analysis: The narrow peak at 169.17°C for pure EZT infers presence of crystalline from of EZT. No representative peaks for drug were observed for SNEP indicating the transformation of crystalline structure of EZT as it may be present in amorphous or molecularly dissolved state in self nanoemulsifying Powder [Figure 4 (a & b)].
In-vitro drug release study: In vitro dissolution studies were performed for SNEP and compared with optimized LS1 liquid SNEDDS formulation. As the emulsification time is below 26 sec, the maximum percentage of the drug release 93% was found to be at the end of 15 min, while plain drug showed only 36% dissolution at the end of 15 min. The drug dissolution study also indicates that the self nanoemulsifying property of the formulation remains unaffected by the conversion of the formulation to solid dosage form.
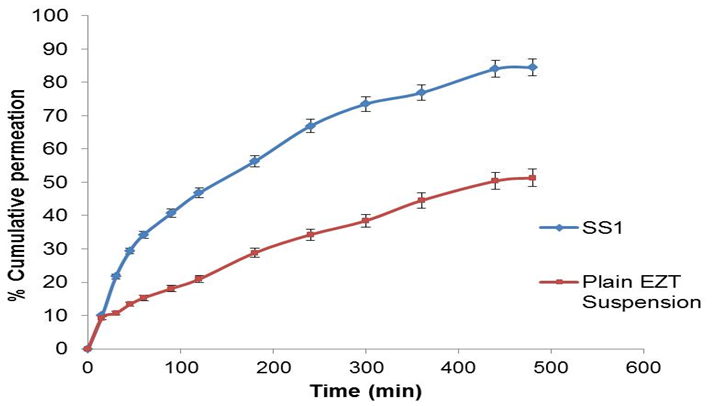
Ex vivo intestinal permeability study: The results of the ex vivo intestinal permeability study are shown in Figure 5[15]. After 8 h of diffusion, 84.50% of the drug was diffused from the SNEP, while from plain drug suspension the diffusion was found to be 51.32%. Thus, the amount of the drug diffused through the biological membrane has near to doubled when it is given in the form of a SNEP. The enhanced diffusion may be explained in terms of (1) the huge specific surface area of the nanoemulsion droplets (2) improved permeation of the EZT because of the presence of surfactant, which reduces the interfacial tension of formulation.
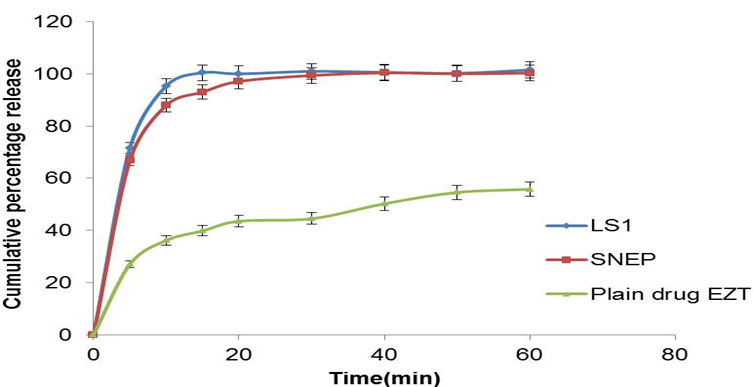
Figure 5: Comparison of dissolution profile of SNEP, optimized SNEDDS formulation and pure drug (Mean ± SD; n = 3)
Short term stability study: There were no significant difference in the dissolution profiles of Ezetimibe in both initial and formulation after 1 month through the stability study period under the different storage conditions. The similarity factor of batch SS1 was found 78.16. This indicates the stability of batch SS1 formulation and its release properties.
Conclusion
From this study it was concluded that SNEDDS of ezetimibe can be successfully prepared by using optimum concentration of 30% Acrysol EL 135, 35% Tween 80 and 35% Transcutol P. It also concluded that Fujicalin exhibited good adsorption property and efficiently formulates SNEP which enhance solubility, dissolution rate and intestinal permeability of ezetimibe.
Acknowledgments
The authors are very thankful to Astron Research Lab. (India), Abitec Corporation (USA), Gattefosse (France), Fuji Chemicals (Japan) and Corel Pharma (India) for providing gift samples.
References
- 1. Sprunk, A., Strachan, C., Grafa, A. Rational formulation development and in vitro assessment of SNEDDS for oral delivery of poorly water soluble drugs. (2012) Eur J Pharm Sci 46(5): 508–515.
- 2. Agrawal, S., Giri, T., Tripathi, D., et al. A review on novel therapeutic strategies for the enhancement of solubility for hydrophobic drugs through lipid and surfactant based SNEDDS: A novel approach. (2012) Ame J Drug Disc Dev 2(4): 143-183.
- 3. Kosoglou, T., Statkevich, P., Levonas, A., et al. Ezetimibe: A review of its metabolism, pharmacokinetics and drug interactions. (2005) Clin Pharmacokinet 44(5): 467-494.
- 4. Patrick, J.E., Kosoglou, T., Stauber, K.L., et al. Disposition of the selective cholesterol absorption inhibitor EZT in healthy male subjects. (2002) Drug Metab Dispo 30(4): 430–437.
- 5. Constantinides, P.P. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. (1995) Pharm Res 12(11): 1561-1572.
- 6. Shah, N., Carvajal, M., Patel, C., et al. Self-emulsifying drug delivery systems (SEDDS) with polyglycolysed glycerides for improving invitro dissolution and oral absorption of lipophilic drugs.(1994) Int J Pharm 106: 15-23.
- 7. Yi, T., Wan, J., Xu, H., et al. A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. (2008) Eur J Pharm Biopharm., 70(2): 439-444.
- 8. Dixit, R.P., Nagarsenker, M.S. Self nanoemulsifying granules of ezetimibe: design, optimization and evaluation. (2008) Eur J Pharm Sci 35(3): 183-192.
- 9. Date, A.A., Nagarsenker, M.S. Design and evaluation of self nanoemulsified drug delivery systems (SNEDDS) for Cefpodoxime Proxetil. (2007) Int J Pharm 329(1-2): 166-172.
- 10. Sheikh, S., Shakeel, S., Talegaonkar, S., et al. Formulation development and optimization using nanoemulsion technique: A Technical note. (2007) APS pharm Sci Tech 8(2): E12-E17.
- 11. Zhang, P., Liu, Y., Feng, N., et al. Preparation and evaluation of self microemulsifying drug delivery system of oridonin. (2008) Int J Pharm 355(1-2): 269-276.
- 12. Bali, V., Ali, M., Ali, J. Study of surfactant combinations and development of a novel nanoemulsion for minimising variations in bioavailability of EZT. (2010) J Drug Target 18(7): 506-519.
- 13. Patel, H.K., Patel, P.V., Misan, C.K., et al. Development and characterization of liquid and solid self-microemulsifying drug delivery system of Tacrolimus. (2012) Asian J Pharm 6(3): 204-211.
- 14. Durgacharan, A.B., D'Souza, J. Formulation and evaluation of solid self-micro emulsifying drug delivery system using aerosil 200 as solid carrier. (2012) Int Curr Pharm J 1(12): 414-419.
- 15. Bandyopadhyay, S., Katare, O., Singh, B. Optimized self nano-emulsifying systems of EZT with enhanced bioavailability potential using long chain and medium chain triglycerides. (2012) Colloids and Surf B Biointerfaces 100: 50-61.
- 16. Kimura, M., Shizuki, M., Miyoshi, K., et al. Relationship between the molecular structures and emulsification properties of edible oils. (1994) Biosci Biotech Biochem 58: 1258-1261.
- 17. Hauss, D.J. Oral 1ipid-basod formu1ations. (2007) Adv Drug De1ivery Rev 59(7): 667-676.
- 18. Chen, M.L. Lipid excipients and delivery systems for pharmaceutical development: A regulatory perspective. (2008) Adv Drug Deliv Rev 60(6): 768-777.
- 19. Patel, A.R., Vavia, P.R. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. (2007) AAPS pharm Sci Tech 9(3): 334-352.
- 20. Constanitinides, P.P., Scalart, J.P., Lancaster, C., et al. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium chain glycerides. (1994) Pharm. Res 11: 1385-1390.










